
Quão eficaz é a terapia genética ELEVIDYS (delandistrogene moxeparvovec-rokl), que foi introduzida no mercado farmacêutico, em relação ao seu custo de 3 milhões de dólares americanos? A revisão dos ensaios clínicos da terapia genética Elevidys será o tópico principal deste ensaio. Avaliaremos o custo e a eficácia desta terapia genética, que foi desenvolvida para tratar a distrofia muscular de Duchenne. O custo do Elevidys vale a pena em comparação aos ensaios clínicos?
Índice
Visão geral dos ensaios clínicos
A Food and Drug Administration (FDA) autorizou uma terapia genética revolucionária em 2023 e 2024 para a distrofia muscular de Duchenne, uma doença rara, mas fatal, de início na infância.

Somente crianças de 4 a 5 anos que pudessem andar (ambular) eram elegíveis para a permissão de 2023.
A decisão de 2024 ampliou essa autorização para cobrir a aprovação convencional para pacientes ambulatoriais com a doença e uma mutação verificada no gene DMD que tenham 4 anos de idade ou mais.
Além disso, o FDA estabeleceu aprovação acelerada para pessoas com 4 anos de idade ou mais que dependem de cadeiras de rodas.
Nenhuma das aprovações foi apoiada por resultados de ensaios clínicos apropriados mostrando que crianças com DMD mantiveram ou restauraram a capacidade motora bruta, nem por avaliações científicas positivas do FDA.
Um diretor do FDA desconsiderou a equipe científica da agência, resultando em ambas as aprovações.
As duas aprovações mal pensadas são discutidas neste artigo, juntamente com as razões pelas quais os líderes do FDA não devem seguir seu padrão no futuro. Ele também oferece sugestões sobre como o FDA pode alterar a maneira como lida com decisões regulatórias contenciosas.
Como o Elevidys foi aprovado pelo FDA
Os dois ensaios clínicos randomizados de Fase 3 do delandistrogene moxeparvovec-rokl (Elevidys) como cura de substituição genética para distrofia muscular de Duchenne (DMD) foram relatados na Nature Medicine em 29 de outubro de 2024. [1] A eficácia clínica deste medicamento não foi comprovada conclusivamente por dois motivos, de acordo com o relatório.
Ambos os ensaios não atingiram seus endpoints primários para benefício clínico. O benefício clínico foi avaliado usando uma escala padronizada que mede a trajetória do desempenho motor bruto, incluindo ficar em pé, andar, pular e levantar a cabeça.
Em segundo lugar, desfechos alternativos que eram marginalmente favoráveis — finalmente empregados pela Food and Drug Administration (FDA) para apoiar a aprovação — não puderam ser verificados por meio de testes estatísticos.
Além disso, o principal resultado alternativo foram os níveis de proteína microdistrofina, um biomarcador que recentemente não mostrou relevância clínica em outro estudo de terapia genética para DMD. [2]
Quando o Elevidys foi aprovado?
Apesar das evidências clínicas limitadas, em 22 de junho de 2023, o delandistrogene moxeparvovec-rokl recebeu a aprovação FDA como a primeira terapia de substituição genética para distrofia muscular de Duchenne (DMD), especificamente para crianças ambulatoriais de 4 a 5 anos. [3]
A aprovação foi obtida por meio do programa de aprovação acelerada do FDA. [4]
Em 20 de junho de 2024, o FDA ampliou sua aprovação para abranger a autorização tradicional para indivíduos ambulatoriais com 4 anos ou mais diagnosticados com a doença e que possuem uma mutação confirmada no gene DMD. A agência também concedeu aprovação acelerada para indivíduos com 4 anos ou mais que são dependentes de cadeiras de rodas. [5]
Uma declaração preocupante foi observada no documento de revisão clínica do FDA referente à ação de aprovação expandida: [6]
O Dr. Peter Marks, Diretor do Centro de Avaliação e Pesquisa Biológica (CBER), aprovou o pedido, anulando a recomendação da equipe de revisão.
O delandistrogene moxeparvovec-rokl agora está aprovado para comercialização nos Estados Unidos. A terapia foi recentemente precificada em mais de $3 milhões por cura. [7]
A Distrofia Muscular de Duchenne (DMD) é uma condição rara, progressiva e fatal, caracterizada pela escassez de alternativas de cura eficazes.
Mutações da Distrofia Muscular de Duchenne (DMD) são primariamente recessivas e ligadas ao X, resultando em um impacto predominante em homens. A incidência de nascimento em homens é de aproximadamente 1 em 3.600. [Leia mais: O que é DMD?]
Essas mutações resultam na degradação do músculo esquelético e cardíaco, que se manifesta na primeira infância, apesar do desenvolvimento musculoesquelético normal. Indivíduos com um genótipo DMD geralmente perdem a deambulação no início da adolescência e têm uma expectativa de vida que geralmente termina na casa dos trinta.
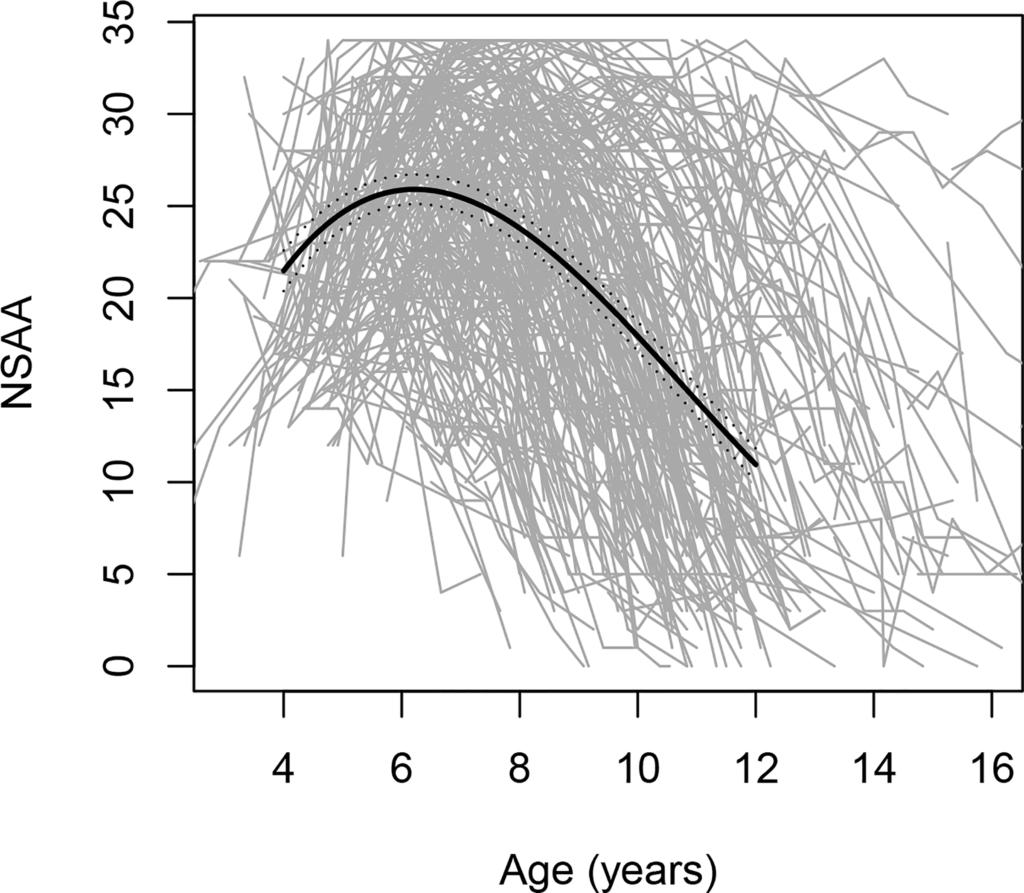
O curso da doença é heterogêneo, como evidenciado pelos dados do FDA que indicam variabilidade significativa nas tendências da função motora entre meninos com DMD de 4 a 16 anos (veja a figura).
Figura. Alterações nas pontuações da North Star Ambulatory Assessment (NSAA), uma escala de desempenho motor bruto, com a idade; inclui 395 trajetórias individuais, cada uma representada por uma linha cinza.
Curas DMD de Sarepta
Delandistrogene moxeparvovec-rokl representa a terapia inaugural modificadora da doença para a maioria dos casos de distrofia muscular de Duchenne (DMD); no entanto, o FDA havia sancionado anteriormente quatro medicamentos para variantes menos comuns de DMD.
Eteplirsen (Exondys 51) é um medicamento sob análise, projetado para tratar mutações específicas de DMD por meio do uso de oligonucleotídeos antisense que permitem o “pulo de exon” direcionado, ignorando assim regiões mutadas na expressão de gene para proteína. [8]
Existem cinco curas aprovadas pelo FDA para DMD, incluindo delandistrogene moxeparvovec-rokl, todas as quais utilizam formas truncadas da proteína muscular essencial distrofina (conforme detalhado abaixo). [9]
Todas essas curas receberam aprovação por meio do programa de aprovação acelerada do FDA. Apesar de serem utilizadas em pacientes por vários anos, a maioria dessas curas não demonstrou vantagens clínicas significativas sobre o placebo.
Além disso, quatro das curas, incluindo o delandistrogene moxeparvovec-rokl, são produzidas por uma única empresa, a Sarepta Therapeutics. [10]
O Dr. Robert Califf, comissário do FDA durante a aprovação do eteplirsen e do delandistrogene moxeparvovec-rokl, expressou preocupações contínuas em relação aos produtos DMD da Sarepta em 2024, afirmando: “Sarepta é como uma maldição para mim”. [11]
Mecanismo de ação do Elevidys
A expressão de um gene que codifica a proteína distrofina é significativamente reduzida ou silenciada em mutações que causam DMD.
Muitos componentes bioquímicos da proteína atravessam as membranas das células musculares. Além disso, ela serve como um amortecedor, mantendo os músculos fortes diante da deterioração potencial causada por ciclos frequentes de contração-expansão. [12]
Teoricamente, a DMD pode ser diminuída ou mesmo revertida ao encorajar a expressão da distrofina nas células musculares daqueles que a têm. O delandistrogene moxeparvovec-rokl, cujo componente ativo é uma sequência de RNA modificada que codifica uma variante encurtada da distrofina conhecida como “micro-distrofina”, foi desenvolvido em resposta a essa noção.
A microdistrofina tem cerca de 30% do tamanho da distrofina não mutada. A proteína encurtada observada em pacientes com o tipo mais brando de distrofia muscular de Becker serviu de base para a invenção da microdistrofina codificada pelo delandistrogene moxeparvovec-rokl.
Como a sequência de RNA teve que ser agrupada em capsídeos virais para ser entregue ao corpo, o gene da distrofina teve que ser truncado. O RNA completo da distrofina ultrapassa o limite de capacidade desses capsídeos.
O delandistrogene moxeparvovec-rokl foi criado usando uma técnica complexa de bioengenharia que resulta em uma proteína que, no máximo, restauraria apenas parcialmente a função da distrofina em todo o corpo. A eficácia dessa terapia intrincada ainda não foi demonstrada em ensaios clínicos.
Aprovação Acelerada de 2023
Quarenta e uma crianças ambulatoriais com DMD, com idades entre 4 e 7 anos, participaram do estudo principal para a primeira aprovação do delandistrogene moxeparvovec-rokl. Elas foram randomizadas 1:1 para receber o medicamento ou um placebo.
O principal resultado clínico predeterminado foi a mudança no desempenho motor bruto ao longo de um período de 48 meses, conforme medido por uma escala padronizada de 17 itens; os pacientes tratados com delandistrogene moxeparvovec-rokl não melhoraram significativamente seu desempenho em comparação ao grupo de controle. [13]
Com base no desfecho substituto da expressão de microdistrofina no tecido muscular 12 semanas após a cura por terapia genética, a Sarepta, a patrocinadora, solicitou aprovação rápida devido ao resultado desfavorável do ensaio clínico.
Apesar do fato de que a expressão de microdistrofina foi notada em 12 semanas, não houve correlação entre os níveis de microdistrofina e a função muscular resultante. Consequentemente, os revisores científicos do FDA chegaram à conclusão de que não houve benefício terapêutico no tratamento de DMD do ensaio delandistrogene moxeparvovec-rokl. De acordo com a revisão clínica do FDA de 2023: [14]
Portanto, mesmo para uma população pequena, como pacientes ambulatoriais com DMD de 4 a 5 anos que têm uma mutação DMD confirmada em seu gene DMD, esses dados são insuficientes para dar suporte à expressão da microdistrofina ELEVIDYS como um desfecho substituto “razoavelmente provável de prever benefício clínico” para a Aprovação Acelerada do ELEVIDYS.
O diretor do Centro de Avaliação e Pesquisa Biológica do FDA desconsiderou os revisores científicos da agência e decidiu conceder aprovação rápida para crianças ambulatoriais de 4 e 5 anos com DMD, apesar dessas duras críticas.
Dados de 8 dos 41 pacientes serviram como base primária para a decisão. Além disso, o patrocinador foi “obrigado como condição de aprovação acelerada” a concluir um segundo ensaio randomizado, cujos resultados eram esperados para o outono de 2023, de acordo com a decisão. [15]
Aprovação expandida em 2024
Sarepta disse em um comunicado à imprensa datado de 30 de outubro de 2023, que o desfecho primário do segundo estudo randomizado fundamental do delandistrogene moxeparvovec-rokl não foi atingido.
Nesse estudo controlado por placebo, 125 pacientes entre 4 e 7 anos foram designados aleatoriamente 1:1 para receber um placebo ou o delandistrogene moxeparvovec-rokl. [16]
A mudança de 52 semanas na mesma pontuação da escala de função muscular usada no experimento anterior foi o principal ponto final. Apenas uma diferença média de 0,65 ponto entre o medicamento e o placebo na escala funcional motora bruta padronizada (que tem uma faixa máxima de 34 pontos) foi encontrada, tornando os resultados não significativos (p=0,25).
O FDA deveria ter parado de aprovar o delandistrogene moxeparvovec-rokl ali. Para a aprovação de 2023, o fabricante enfatizou, em vez disso, os resultados secundários que eles disseram que produziram “resultados robustos e estatisticamente significativos… [que] apoiam um suplemento de eficácia”.
Os revisores científicos do FDA foram severamente desdenhosos dessas novas descobertas. Em particular, os testes de caminhada/corrida de 10 metros e o tempo que levou para se levantar de uma posição no chão foram dois resultados secundários importantes.
Os indivíduos que tomaram delandistrogene moxeparvovec-rokl realizaram essas atividades em média 0,5 segundos mais rápido do que os indivíduos que tomaram placebo, que levaram 3,5 e 4,9 segundos no início do estudo, respectivamente.
Essas descobertas, que não foram pré-especificadas nem corrigidas estatisticamente para as análises de dados múltiplos, “não podem apoiar a eficácia do ELEVIDYS”, de acordo com os revisores do FDA.
Os dados, de acordo com os revisores do FDA, também eram “enganosos e não podem orientar nenhuma parte interessada — incluindo pacientes, familiares e cuidadores, e prescritores — na tomada de decisões informadas sobre os benefícios potenciais da cura com ELEVIDYS”. [17]
Entretanto, em junho de 2024, o diretor do CBER anulou sua equipe e aprovou o delandistrogene moxeparvovec-rokl pela segunda vez, apesar das avaliações desfavoráveis dos cientistas do FDA. [18]
Conforme mencionado anteriormente, a segunda aprovação expandiu e fortaleceu a permissão para incluir a aprovação típica em pacientes ambulatoriais com a condição que tenham 4 anos de idade ou mais e tenham uma mutação verificada no gene DMD. Além disso, a permissão acelerada para pessoas dependentes de cadeira de rodas com 4 anos ou mais foi incorporada na segunda aprovação. [19]
A indicação substituta da conexão entre os níveis de proteína microdistrofina e os dados do tempo para levantar após ficar deitado no chão foi um fator importante no memorando de decisão do Diretor do CBER de 2024.
Essa ligação, no entanto, não foi visualmente (graficamente) convincente nem estatisticamente significativa (p=0,1388) devido à inclinação suave (-0,012 segundos para cada alteração percentual no nível de microdistrofina do músculo).
Além disso, um grande número de pontos de dados desviou significativamente da linha de regressão estimada.
Como apenas 25% dos indivíduos no segundo ensaio principal foram incluídos nesta análise, os especialistas do FDA notaram explicitamente que os resultados devem ser “interpretados com cautela” ao avaliar os dados de dose-resposta da microdistrofina. Como resultado, apesar da justificativa científica para usar a inserção do gene da microdistrofina para tratar DMD, os ensaios clínicos não mostraram que a terapia genética tenha quaisquer efeitos positivos. [20]
De fato, “quantidades significativas” de expressão de microdistrofina foram vistas nos músculos de pacientes com DMD em um recente ensaio de Fase 3 de uma terapia genética para DMD desenvolvida pela Pfizer, mas não houve melhora clínica proporcional. De acordo com relatos, a Pfizer parou de trabalhar em sua cura. [21]
Comentários da família
Preocupações semelhantes foram expressas por aqueles que são diretamente impactados pela DMD. Preocupações quanto à segurança e eficácia do delandistrogene moxeparvovec-rokl levaram um pai a escrever em setembro de 2024 que eles tinham escolhido não buscar terapia para seu filho. [22]
Em um vídeo online, um pai diferente criticou Sarepta por criar uma cura que era insuficiente e logo se tornaria obsoleta. Em resposta, um grupo de apoio a pais que estava hospedando o vídeo foi acusado de remover a crítica em vídeo delandistrogene moxeparvovec-rokl de seu site depois que Sarepta Therapeutics ameaçou parar de financiar o grupo. [23]
O custo do Elevidys vale a pena em comparação aos ensaios clínicos?
Considerando os resultados dos ensaios clínicos, o preço de 3 milhões de dólares para a terapia genética Elevidys é razoável? [Leia mais: Distrofia Muscular de Duchenne: Tratamento e Custo]
Considerando a quantidade de distrofina produzida no corpo após o uso de Elevidys e as pontuações da North Star Ambulatory Assessment (NSAA) examinadas em ensaios clínicos, 3 milhões de dólares é muito exorbitante para esse método de tratamento.
Nosso apelo à Sarepta, desenvolvedora do Elevidys, e à Roche, autoridade global em marketing: Reduza imediatamente esse alto preço definido para essa finalidade comercial!
Saber mais: Possíveis novas terapias genéticas futuras para a distrofia muscular de Duchenne
Primeiro Anúncio de Morte
Em 18 de março de 2025, Sarepta Therapeutics disse que a primeira fatalidade registrada associada à sua terapia genética para distrofia muscular de Duchenne, Elevidys, ocorreu em um garoto de 16 anos. [Ler mais]
Conclusão
Um diretor do centro anulou a decisão da equipe científica do FDA, que tinha a tarefa de avaliar o medicamento, e o FDA aprovou o delandistrogene moxeparvovec-rokl como terapia genética para DMD.
Os dois ensaios principais do delandistrogene moxeparvovec-rokl falharam em atingir seus principais objetivos clínicos. Indicadores usados para resgatar a aplicação do delandistrogene moxeparvovec-rokl, como endpoints secundários (por exemplo, testes de caminhada/corrida de 10 metros) e o indicador substituto indireto dos níveis de microdistrofina, eram problemáticos por si só.
Como resultado, acreditamos que a liderança do FDA criou um precedente arriscado que não protege o público de ser exposto a um medicamento ineficaz, caro e fisicamente exigente. [24]
Saber mais: Mercado de distrofia muscular de Duchenne cresce: mas nem todas as famílias têm acesso aos tratamentos
Uma maneira de reagir à decisão do FDA é dizer que critérios de aprovação fracos são o problema e que padrões de aprovação mais rigorosos e procedimentos pós-comercialização são a solução. [25]
Os processos internos do FDA devem ser alterados para tornar muito mais difícil para funcionários individuais em funções de liderança anular a avaliação consensual dos cientistas da agência designados para uma decisão regulatória, embora concordemos que padrões de aprovação e requisitos pós-comercialização mais rigorosos sejam necessários.
De acordo com Califf, o comissário do FDA na época das duas decisões de aprovação do delandistrogene moxeparvovec-rokl, aqueles que se opõem à decisão têm uma “visão muito simplista das evidências clínicas” sobre as dificuldades no desenvolvimento de terapias para distúrbios graves e incomuns. [26]
Não concordamos com essa avaliação. O FDA deve aprimorar seu processo de tomada de decisão para novos fármacos, sejam eles para doenças comuns ou incomuns, como resultado da aprovação do delandistrogene moxeparvovec-rokl.
Saber mais: Representante turco da DMD WarrioR compartilha suas opiniões sobre a terapia genética Elevidys
Em uma entrevista antes de sair, Califf também discutiu a decisão, apontando que famílias com doenças tão graves e incomuns estavam compreensivelmente ansiando por qualquer esperança, e que, como um nomeado político, ele também estava relutante em anular as decisões tomadas pelos funcionários de carreira do FDA. Embora esses fatores sejam significativos, eles não são suficientes para apoiar a decisão do FDA de aprovar o delandistrogene moxeparvovec-rokl. [27]

{kind=link}