









 FDA en 2019")
La Food and Drug Administration a publié plus de 200 lettres qu'elle a envoyées aux entreprises après le rejet de leurs médicaments jeudi, attirant l'attention sur une partie parfois négligée du processus d'examen des médicaments. L'une de ces lettres de rejet a été envoyée pour Vyondys 53 (golodirsen), un médicament fabriqué par Sarepta Therapeutics et développé pour la thérapie de saut d'exon 53.
L'agence s'est concentrée uniquement sur les lettres adressées aux fabricants dont les produits ont finalement été approuvés, dont la plupart avaient déjà été commercialisés au fil des ans. En regroupant toutes ces lettres en un seul endroit, l'agence a affirmé que cette activité s'inscrivait dans un effort plus large de promotion de la transparence.

Table des matières
Détails de la lettre de rejet FDA concernant Vyondys 53
Le FDA a émis sa lettre de rejet de 2019 pour Vyondys 53 (golodirsen), un traitement de saut d'exon 53 pour la dystrophie musculaire de Duchenne. Le FDA a déclaré qu'étant donné la « faible augmentation des niveaux de dystrophine raccourcie » fournie par le traitement, le bénéfice clinique de Vyondys 53 serait probablement « proportionnellement faible ».
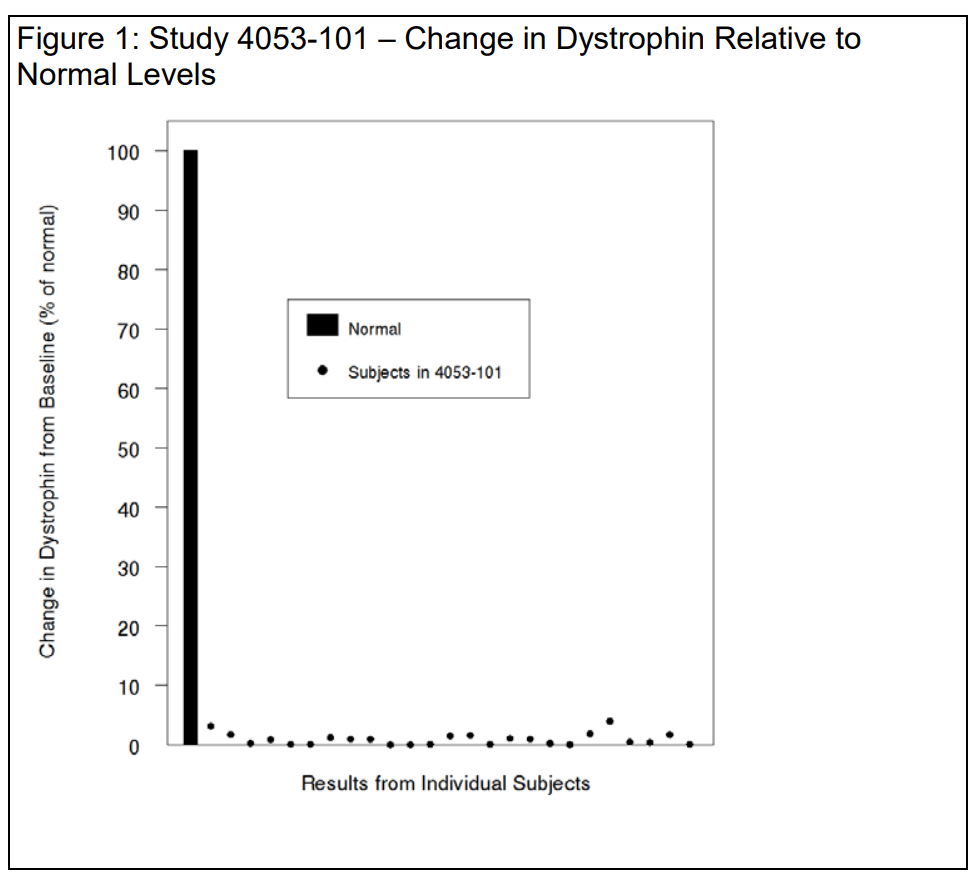
Le taux de dystrophine a augmenté de 9 parties pour mille
La lettre de rejet du FDA pour Vyondys 53 (golodirsen) a noté une très faible augmentation des niveaux de dystrophine après l'utilisation du médicament:
Pour les 25 patients évaluables de l'étude, le taux moyen de dystrophine initial, mesuré par Western blot, était de 0,10 ± 0,07 (pourcentage de la normale ; moyenne ± écart-type). À la semaine 48, le taux moyen de dystrophine était de 1,02 ± 1,03 (pourcentage de la normale ; moyenne ± écart-type), ce qui correspond à une augmentation moyenne de 0,92 ± 1,01 pour cent de la normale.
Les variations individuelles de la dystrophine sont présentées ci-dessous dans la figure 1, à l'aide d'une échelle normale (100%) afin de mettre les données en perspective et d'éviter une exagération de l'ampleur de l'effet. Vous constatez que les augmentations moyennes de la dystrophine tronquée sont similaires en réponse au golodirsen et à l'eteplirsen chez les patients présentant des mutations sensibles au saut de l'exon 53 et de l'exon 51, respectivement – avec des augmentations moyennes absolues de 0,9%, soit 9 pour mille. Nous sommes d'accord.
Si l’on accepte l’hypothèse selon laquelle une petite augmentation de la dystrophine tronquée, de l’ordre de 9 parties pour mille, est raisonnablement susceptible de prédire un bénéfice clinique, il semble raisonnable de supposer que le bénéfice clinique serait proportionnellement faible.
Effet clinique du Vyondys 53
Les données de marche de 6 minutes de l'étude 4053-101 ne sont pas abordées ici, mais elles montrent une perte progressive des fonctions physiques chez la quasi-totalité des garçons. De plus, il n'existe aucune corrélation entre le maintien des performances physiques et l'ampleur de la production de dystrophine tronquée, ce qui suggère que, si le golodirsen a bien un effet clinique, son ampleur est faible.
Effets secondaires graves du Vyondys 53, selon le FDA
En décembre 2019, le FDA a autorisé le Vyondys 53 de Sarepta comme première option thérapeutique ciblée pour les patients atteints de DMD susceptibles de sauter l'exon 53. Cependant, le médicament avait été rejeté par le FDA quatre mois plus tôt dans une lettre de recommandation signée par Ellis Unger, alors responsable du Bureau d'évaluation des médicaments du Centre d'évaluation et de recherche en médecine.
Dans sa justification du rejet, le FDA a indiqué que, compte tenu de la « faible augmentation de la dystrophine tronquée » provoquée par le traitement, l'effet thérapeutique du Vyondys 53 serait probablement « proportionnellement faible ». Unger a poursuivi en expliquant qu'il n'existait « aucune corrélation » entre la quantité de dystrophine produite par le Vyondys 53 chez les garçons et leurs performances physiques, et que « pratiquement tous » les garçons examinés après avoir reçu le médicament présentaient une perte croissante de leurs fonctions physiques.
Comme si les bleus ne suffisaient pas, Unger a énuméré une longue liste de problèmes de sécurité auxquels les utilisateurs de Vyondys 53 devaient faire face. Parmi ceux-ci figuraient la « toxicité rénale » et les « infections graves liées à l'administration du médicament ». Il affirme que ces deux « risques » sont potentiellement mortels, ces dernières étant « difficiles, voire impossibles à surveiller ».
Compte tenu des deux décès récents dus à une insuffisance hépatique sévère liés à la thérapie génique Elevidys de Sarepta, ce deuxième avertissement est particulièrement préoccupant. Comme l'a souligné Unger dans sa lettre, les deux voies d'administration ont été associées à des lésions hépatiques, même si Vyondys 53 est un oligonucléotide antisens (ASO), contrairement à une thérapie génique basée sur un virus adéno-associé (AAV) comme Elevidys.
En savoir plus: Lettre de refus Vyondys 53 FDA
Comment Vyondys 53 a-t-il été approuvé ?
Afin de répondre aux préoccupations soulevées par l'organisme de réglementation dans la CRL, Sarepta a ensuite interjeté appel et rencontré le FDA. Cette décision a finalement abouti à l'approbation du Vyondys 53 par Peter Stein, alors directeur du Bureau des nouveaux médicaments du FDA et qui a quitté l'agence en avril.
Les sociétés pharmaceutiques ne publient pas toutes les informations
« Pendant trop longtemps, les développeurs de médicaments ont joué aux devinettes en s'y prenant dans le cadre du FDA », a déclaré le commissaire du FDA, Marty Makary, MD, MPH. « Les développeurs de médicaments et les marchés financiers recherchent tous deux la prévisibilité. Aujourd'hui, nous sommes sur le point de la leur offrir, avec pour objectif ultime de mettre plus rapidement à la disposition des patients des traitements efficaces et des remèdes. »
Étant donné que le FDA s'est traditionnellement abstenu de publier les LCR des demandes en attente, les promoteurs déforment souvent les raisons de sa décision auprès de leurs parties prenantes et du public. Selon une analyse réalisée en 2015 par les chercheurs du FDA, les promoteurs ont évité de mentionner 85% des préoccupations du FDA concernant la sécurité et l'efficacité lors de l'annonce publique du rejet de leur demande. De plus, lorsque le FDA exige un nouvel essai clinique pour évaluer la sécurité ou l'efficacité, cette information cruciale n'est pas divulguée dans environ 40% des cas. Les enseignements tirés des refus d'approbation ne sont pas non plus partagés au sein de l'industrie, ce qui conduit les entreprises à répéter les mêmes erreurs. – En savoir plus : FDA adopte une transparence radicale en publiant des lettres de réponse complètes –

{kind=link}