









 FDA em 2019")
A Food and Drug Administration publicou mais de 200 cartas enviadas às empresas depois que seus medicamentos foram rejeitados na quinta-feira, chamando a atenção para uma parte às vezes esquecida do processo de revisão de medicamentos. Uma dessas cartas de rejeição foi enviada para Vyondys 53 (golodirsen), um medicamento fabricado pela Sarepta Therapeutics e desenvolvido para terapia de salto do exon 53.
A agência se concentrou apenas em cartas enviadas aos fabricantes cujos produtos foram eventualmente aprovados, a maioria das quais já havia sido lançada ao longo dos anos. Ao reunir todas as cartas em um só lugar, a agência alegou que a atividade era parte de um esforço mais amplo para promover a transparência.

Índice
Detalhes da Carta de Rejeição FDA sobre Vyondys 53
O FDA emitiu sua carta de rejeição de 2019 para Vyondys 53 (golodirsen), um tratamento que ignora o exon 53 para distrofia muscular de Duchenne. O FDA afirmou que, dado o “pequeno aumento nos níveis de distrofina encurtada” fornecido pelo tratamento, o benefício clínico do Vyondys 53 provavelmente seria “comparavelmente pequeno”.
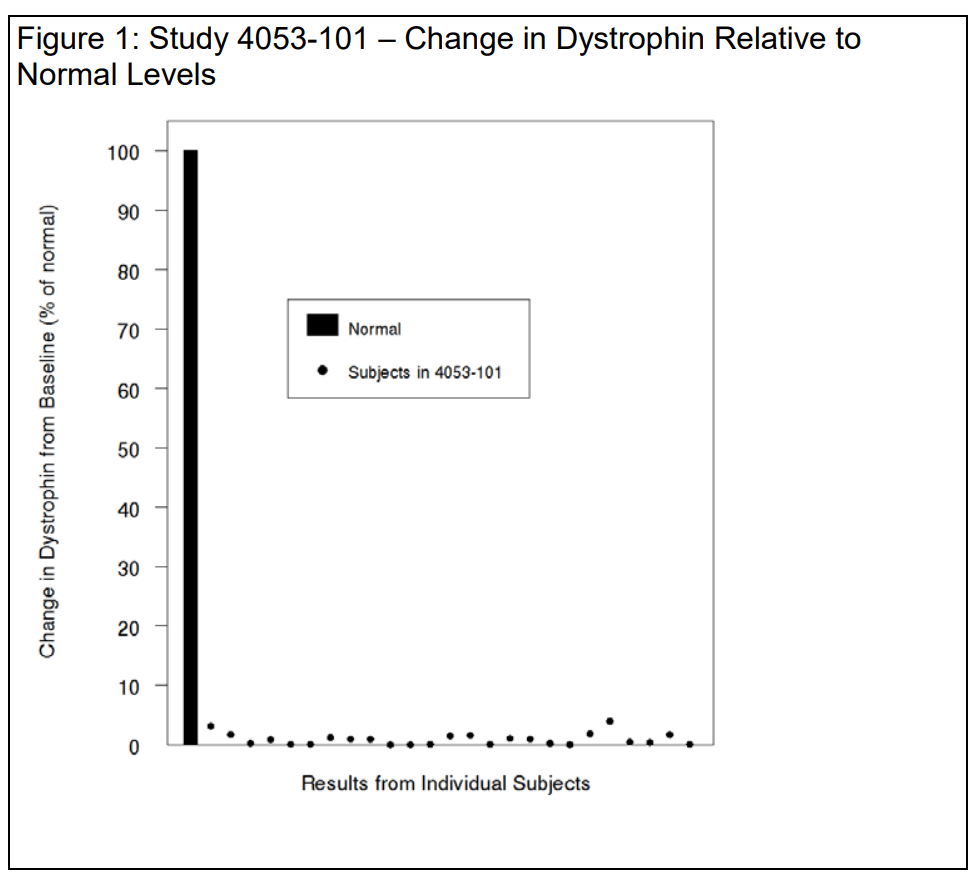
O nível de distrofina aumentou 9 partes em mil
A carta de rejeição do FDA para Vyondys 53 (golodirsen) observou um aumento muito pequeno nos níveis de distrofina após o uso do medicamento:
Para os 25 pacientes avaliáveis no estudo, o nível médio basal de distrofina avaliado por Western blot foi de 0,10 ± 0,07 (porcentagem do normal; média ± DP). Na semana 48, o nível médio de distrofina foi de 1,02 ± 1,03 (porcentagem do normal; média ± DP), correspondendo a um aumento médio de 0,92 ± 1,01% do normal.
As alterações individuais na distrofina são mostradas abaixo na Figura 1, utilizando uma escala normal (100%) para situar os dados na perspectiva correta e evitar exageros no tamanho do efeito. Você observa que os aumentos médios na distrofina truncada são semelhantes em resposta a golodirsen e eteplirsen em pacientes com mutações passíveis de salto do exon 53 e do exon 51, respectivamente – com aumentos médios absolutos de 0,9%, ou seja, 9 partes em mil. Concordamos.
Se aceitarmos a premissa de que um pequeno aumento na distrofina truncada, na ordem de 9 partes por mil, tem probabilidade razoável de prever benefício clínico, parece razoável supor que o benefício clínico seria proporcionalmente pequeno.
Efeito clínico do Vyondys 53
Os dados de caminhada de 6 minutos do Estudo 4053-101 não são discutidos aqui, mas mostram perda progressiva da função física em praticamente todos os meninos. Além disso, não há correlação entre a manutenção do desempenho físico e a magnitude da produção truncada de distrofina, sugerindo ainda que, se houver de fato um efeito clínico do golodirsen, o tamanho do efeito é pequeno.
Efeitos colaterais graves do Vyondys 53, de acordo com o FDA
Em dezembro de 2019, o FDA autorizou o Vyondys 53 da Sarepta como a primeira opção terapêutica direcionada para pacientes com DMD suscetíveis ao salto do exon 53. No entanto, o medicamento havia sido rejeitado pelo FDA quatro meses antes, em uma LCR assinada por Ellis Unger, então chefe do Escritório de Avaliação de Medicamentos do Centro de Avaliação e Pesquisa Médica.
O FDA declarou em sua justificativa para a rejeição que, considerando o “pequeno aumento na distrofina truncada” que o tratamento evocou, o efeito terapêutico do Vyondys 53 provavelmente seria “comparavelmente pequeno”. Unger continuou explicando que não havia “nenhuma correlação” entre a quantidade de distrofina que o Vyondys 53 produziu nos meninos e seu desempenho físico, e que “essencialmente todos” os meninos que foram examinados após receber o medicamento apresentaram perda crescente da função física.
Como se os hematomas não bastassem, Unger enumerou uma longa lista de problemas de segurança com os quais os usuários do Vyondys 53 tiveram que lidar. Entre eles, estavam "toxicidade renal" e "infecções graves relacionadas à administração do medicamento". Ele afirma que ambos "têm o potencial de serem fatais", sendo o último "difícil ou impossível de monitorar".
Considerando as duas mortes recentes por insuficiência hepática grave associadas à terapia genética Elevidys da Sarepta, este segundo alerta é especialmente preocupante. Como Unger observou na carta, ambas as vias têm sido associadas a danos hepáticos, embora o Vyondys 53 seja um oligonucleotídeo antisense (ASO), em oposição a uma terapia genética baseada em vírus adeno-associados (AAV), como o Elevidys.
Ler mais: Carta de rejeição Vyondys 53 FDA
Como o Vyondys 53 foi aprovado?
Para abordar as preocupações levantadas pelo regulador no CRL, Sarepta posteriormente entrou com um recurso e se reuniu com o FDA. Isso finalmente resultou na aprovação do Vyondys 53 pelo então chefe do Escritório de Novos Medicamentos do FDA, Peter Stein, que deixou a agência em abril.
As empresas farmacêuticas não publicam todas as informações
“Por muito tempo, os desenvolvedores de medicamentos têm jogado um jogo de adivinhação ao navegar pelo FDA”, disse o Comissário do FDA, Dr. Marty Makary, MPH. “Tanto os desenvolvedores de medicamentos quanto os mercados de capitais querem previsibilidade. Então, hoje estamos um passo mais perto de entregar isso a eles, com o objetivo final de levar curas e tratamentos significativos aos pacientes mais rapidamente.”
Como o FDA historicamente se absteve de publicar LCRs para pedidos pendentes, os patrocinadores frequentemente deturpam a justificativa por trás da decisão do FDA para suas partes interessadas e o público. De acordo com uma análise de 2015 conduzida por pesquisadores do FDA, os patrocinadores evitaram mencionar as preocupações do FDA com relação à segurança e eficácia ao anunciar publicamente que seu pedido não foi aprovado. Além disso, quando o FDA solicita um novo ensaio clínico para fins de segurança ou eficácia, essas informações críticas não são divulgadas em aproximadamente 40% das vezes. As lições aprendidas com as não aprovações também não são compartilhadas com a indústria, levando as empresas a cometerem erros semelhantes repetidamente. – Leia mais: FDA adota transparência radical ao publicar cartas de resposta completas –

{kind=link}