
Le développement d'un médicament est un processus complexe, long et coûteux qui s'étend sur plusieurs années, généralement entre 10 et 15 ans, de la découverte initiale à la mise sur le marché. Y compris pour les traitements contre la dystrophie musculaire de Duchenne, il comprend plusieurs étapes, chacune soigneusement conçue pour évaluer l'innocuité, l'efficacité et les risques potentiels d'un nouveau médicament avant sa mise à disposition au grand public. Les étapes du développement d'un médicament se décomposent en recherche préclinique et essais cliniques, qui constituent le principal objectif des agences réglementaires telles que la Food and Drug Administration (FDA) et l'Agence européenne des médicaments (EMA).
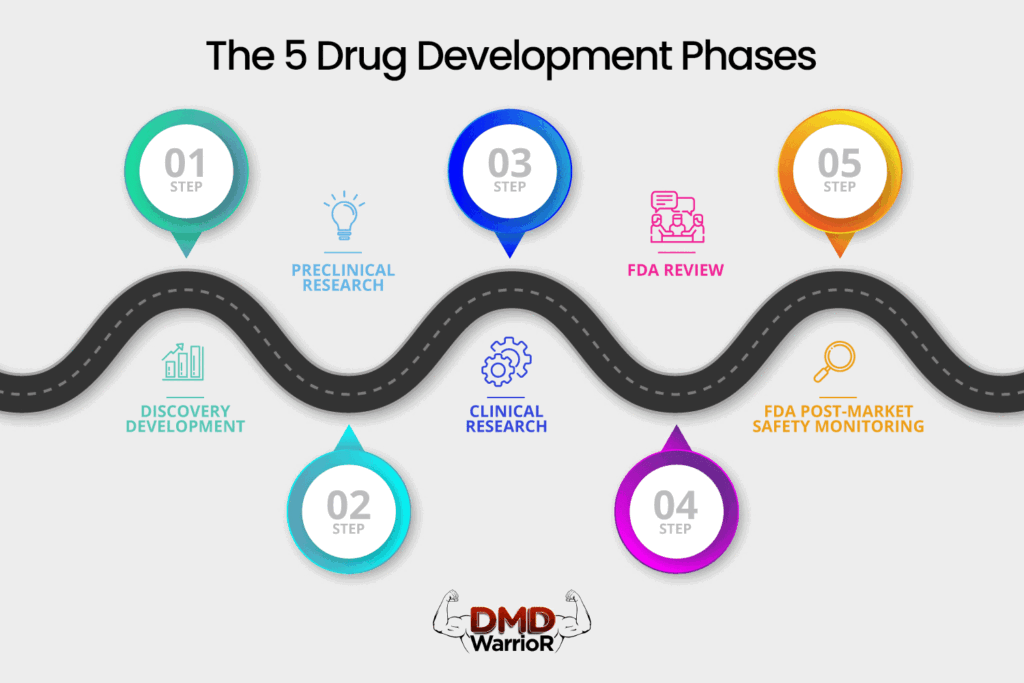
Un nouveau médicament doit passer par cinq étapes distinctes pour être considéré comme un « succès » : 1) Découverte et développement ; 2) Recherche préclinique ; 3) Essais cliniques ; 4) Examen du médicament FDA ; et 5) Surveillance de la sécurité post-commercialisation FDA. [FDA]

Table des matières
Découverte et développement
Avant même qu'un médicament n'entre en phase d'essai clinique, d'importantes recherches préliminaires sont menées. Cette phase comprend :
- Découverte de médicamentsLa première étape consiste à identifier une cible biologique (comme une protéine ou un gène) impliquée dans une maladie. Les scientifiques peuvent analyser de vastes banques de composés afin d'identifier des candidats susceptibles d'agir sur la cible. Ces composés sont ensuite optimisés pour améliorer leur efficacité et réduire les effets secondaires potentiels.
Recherche préclinique
- Préclinique : Une fois qu'un composé prometteur est identifié, il est soumis à des tests en laboratoire in vitro (hors organisme vivant) et in vivo (sur des organismes vivants, généralement des animaux) afin d'évaluer sa toxicité, sa pharmacocinétique (absorption, distribution, métabolisation et excrétion dans l'organisme) et sa pharmacodynamie (effets du médicament sur l'organisme). Ces études permettent de déterminer si le médicament est susceptible d'être sûr et efficace chez l'homme.
- Études toxicologiques : Lors des recherches précliniques, les scientifiques mènent des études détaillées pour évaluer les éventuels effets nocifs du médicament. L'objectif est d'identifier la dose maximale tolérée et les éventuels effets indésirables.
Si les études précliniques sont concluantes, le médicament passe à la phase d’essai clinique, qui consiste à tester le médicament sur l’homme.
Essais cliniques
Que sont les essais cliniques de phase 1, de phase 2 et de phase 3 ?
Le processus d'essai clinique est divisé en quatre phases distinctes (phase 1, phase 2, phase 3 et phase 4) afin d'évaluer progressivement l'innocuité, l'efficacité et le rapport bénéfice/risque global d'un médicament chez l'homme. Chaque phase a un objectif spécifique dans le processus d'évaluation.
Phase 1 : Sécurité et dosage
La phase 1 marque le premier essai du médicament chez l'homme après des recherches précliniques. L'objectif principal des essais de phase 1 est d'évaluer le profil de sécurité du médicament, de déterminer une plage posologique sûre et d'identifier les éventuels effets secondaires.
- Conception de l'étudeEn règle générale, les essais de phase 1 sont de petite envergure, impliquant 20 à 100 volontaires sains ou personnes atteintes de la maladie. Ces essais sont généralement menés dans un environnement clinique contrôlé, comme un hôpital ou un centre de recherche.
- Objectifs:
- Sécurité:Les chercheurs surveillent de près les volontaires pour détecter tout effet indésirable, allant de symptômes légers à des réactions plus graves.
- Dosage:La posologie initiale du médicament est souvent prudente. La dose est progressivement augmentée selon un processus appelé « augmentation de dose » afin de déterminer la dose maximale tolérée.
- Pharmacocinétique et pharmacodynamique:Les chercheurs étudient également la manière dont le médicament est absorbé, distribué, métabolisé et excrété par l’organisme, ainsi que ses effets biologiques.
- DuréeLes essais de phase 1 durent généralement quelques mois. En cas de succès, le médicament passe en phase 2.
Phase 2 : efficacité et effets secondaires
Les essais de phase 2 sont menés pour étudier plus en détail l’efficacité et la sécurité du médicament dans une population de patients spécifique atteinte de la maladie ou de l’affection que le médicament est censé traiter.
- Conception de l'étudeCes essais sont généralement plus importants que la phase 1 et impliquent de 100 à 300 participants. L'objectif est de tester l'efficacité du médicament chez les patients et d'identifier les éventuels effets indésirables associés à une utilisation à long terme.
- Objectifs:
- EfficacitéLes chercheurs cherchent à déterminer si le médicament produit l'effet thérapeutique souhaité chez les patients atteints de la maladie ciblée. L'efficacité est mesurée par des résultats cliniques, tels que l'amélioration des symptômes ou les biomarqueurs de la maladie.
- Sécurité:Comme pour la phase 1, les chercheurs continuent de surveiller la sécurité, mais l’objectif principal est d’identifier les effets secondaires qui pourraient survenir chez les patients atteints de la maladie.
- Dose optimale:Au cours de la phase 2, les chercheurs affinent davantage le schéma posologique du médicament en fonction des informations obtenues lors de la phase 1.
- DuréeLes essais de phase 2 durent généralement de quelques mois à quelques années. Si le médicament présente une efficacité et un profil de sécurité favorable, il passe à la phase 3.
Phase 3 : Essais de confirmation (efficacité et sécurité)
Les essais de phase 3 sont des études à grande échelle visant à confirmer l'efficacité du médicament, à surveiller ses effets secondaires et à le comparer aux traitements existants ou à un placebo. Cette phase joue un rôle crucial pour déterminer si le médicament doit être approuvé par les autorités réglementaires.
- Conception de l'étudeLes essais de phase 3 impliquent un nombre beaucoup plus important de participants, généralement compris entre 1 000 et 3 000, voire plus. Ces essais sont souvent multicentriques, c'est-à-dire menés sur plusieurs sites, voire à l'international.
- Objectifs:
- Efficacité:L’objectif est de confirmer que le médicament apporte un bénéfice cliniquement significatif par rapport aux traitements existants ou à un placebo.
- Sécurité:Les chercheurs continuent de surveiller les effets secondaires, mais la taille plus importante de l’échantillon permet d’identifier les événements indésirables moins courants qui auraient pu être manqués lors des phases précédentes.
- Efficacité comparative:Les essais de phase 3 comparent souvent le nouveau médicament avec des traitements standards ou des placebos pour démontrer sa supériorité ou sa non-infériorité.
- DuréeLes essais de phase 3 peuvent durer plusieurs années en raison de la collecte de données approfondie nécessaire. Ces essais constituent souvent la partie la plus coûteuse et la plus longue du processus de développement d'un médicament.
- Résultat finalSi un essai de phase 3 est concluant, le promoteur du médicament peut soumettre une demande d'autorisation de mise sur le marché (NDA) ou une demande d'autorisation de mise sur le marché (BLA) aux autorités réglementaires, comme les organismes FDA ou EMA. En cas d'approbation, le médicament passe à la phase finale : Phase 4.
Examen du médicament FDA
Les résultats d'innocuité et d'efficacité d'un composé médicamenteux issus d'essais cliniques sont examinés par un panel de spécialistes comprenant des médecins, des chimistes, des statisticiens, des microbiologistes et des pharmacologues une fois qu'il a passé les phases 1, 2, 3 et 4 des essais cliniques.
Une entreprise de biotechnologie ou pharmaceutique doit déposer une demande d'autorisation de mise sur le marché (BLA) pour les produits biologiques ou une demande d'autorisation de mise sur le marché (NDA) pour les médicaments afin de solliciter une évaluation FDA. Ensuite, l'FDA doit approuver la demande et désigner un groupe de professionnels chargé d'en évaluer le bien-fondé.
Le groupe examine l'étude clinique en tenant compte de l'analyse risques-bénéfices du médicament, des effets secondaires possibles et des résultats obtenus par les patients. Le FDA autorise la fabrication, la commercialisation et la distribution du médicament aux États-Unis s'il est jugé sûr et efficace pour l'usage prévu.
Surveillance de la sécurité post-commercialisation du FDA
Une fois qu'un médicament est approuvé par les organismes de réglementation, il entre dans la phase post-commercialisation, également appelée phase 4. Au cours de cette phase, le médicament est disponible pour une utilisation par la population générale, mais les chercheurs continuent de surveiller sa sécurité à long terme, son efficacité et tout effet secondaire rare ou à long terme.
- Surveillance post-commercialisation:En phase 4, les études en cours et les rapports des patients (tels que ceux provenant des systèmes de déclaration des événements indésirables) aident à identifier les effets secondaires rares qui n’auraient pas été détectés lors des essais précédents.
- Données du monde réel:Les chercheurs collectent également des données sur la façon dont le médicament fonctionne dans des contextes réels et auprès de diverses populations qui n’ont peut-être pas été représentées de manière adéquate dans les essais cliniques.
Apprendre encore plus: Les remèdes contre la maladie de Duchenne (liste de toutes les recherches)
Conclusion : L'importance de chaque phase du développement d'un médicament
Chaque phase du développement clinique d'un médicament joue un rôle crucial pour garantir son innocuité et son efficacité avant sa mise sur le marché. De la phase 1, où la sécurité est la préoccupation principale, à la phase 3, qui teste rigoureusement le rapport bénéfice/risque global d'un médicament sur une large population de patients, en passant par la phase 4, qui surveille les effets à long terme après la mise sur le marché, chaque phase contribue à mieux comprendre l'impact du médicament sur les patients.
Le processus global de développement de médicaments est un exercice d'équilibre complexe, où les entreprises pharmaceutiques, les organismes de réglementation et les chercheurs collaborent pour commercialiser des traitements innovants et révolutionnaires, tout en minimisant les risques potentiels. Le temps, les efforts et les coûts investis dans le développement de médicaments sont considérables, mais ils sont essentiels pour faire progresser la science médicale et améliorer les résultats des patients partout dans le monde.

{kind=link}