










Wie wirksam ist die Gentherapie ELEVIDYS (Delandistrogen Moxeparvovec-Rokl), die auf dem Pharmamarkt eingeführt wurde, im Verhältnis zu ihren Kosten von 3 Millionen US-Dollar? Die Überprüfung der klinischen Studien zur Elevidys-Gentherapie wird das Hauptthema dieses Aufsatzes sein. Wir werden die Kosten und die Wirksamkeit dieser Gentherapie bewerten, die zur Behandlung der Duchenne-Muskeldystrophie entwickelt wurde. Lohnen sich die Kosten für Elevidys im Vergleich zu klinischen Studien?
Inhaltsverzeichnis
Übersicht über klinische Studien
Die Food and Drug Administration (FDA) hat für die Jahre 2023 und 2024 eine revolutionäre Gentherapie für die seltene, aber tödliche, im Kindesalter auftretende Krankheit Muskeldystrophie Duchenne zugelassen.

Nur Jugendliche im Alter von 4–5 Jahren, die laufen konnten (ambulant), hatten Anspruch auf die Genehmigung für 2023.
Mit der Entscheidung aus dem Jahr 2024 wurde diese Zulassung erweitert, um auch die konventionelle Zulassung für ambulante Patienten mit der Erkrankung und einer nachgewiesenen Mutation im DMD-Gen abzudecken, die vier Jahre oder älter sind.
Darüber hinaus ermöglicht das FDA eine beschleunigte Zulassung für Personen ab 4 Jahren, die auf einen Rollstuhl angewiesen sind.
Keine der Zulassungen wurde durch entsprechende Ergebnisse klinischer Studien gestützt, die zeigten, dass Kinder mit DMD ihre grobmotorischen Fähigkeiten beibehielten oder wiederherstellten, noch durch positive wissenschaftliche FDA-Bewertungen.
Ein Direktor von FDA missachtete die wissenschaftlichen Mitarbeiter der Agentur, was zu beiden Genehmigungen führte.
In diesem Dokument werden die beiden schlecht durchdachten Genehmigungen sowie die Gründe erörtert, warum die Verantwortlichen von FDA in Zukunft nicht ihrem Beispiel folgen sollten. Es werden auch Vorschläge gemacht, wie FDA seine Art, mit umstrittenen Regulierungsentscheidungen umzugehen, ändern kann.
Wie Elevidys von FDA genehmigt wurde
Die beiden randomisierten klinischen Phase-3-Studien zu Delandistrogen Moxeparvovec-Rokl (Elevidys) als Genersatztherapie für die Duchenne-Muskeldystrophie (DMD) wurden am 29. Oktober 2024 in Nature Medicine veröffentlicht. [1] Die klinische Wirksamkeit dieses Medikaments sei aus zwei Gründen nicht abschließend bewiesen, heißt es in diesem Bericht.
Beide Studien erreichten ihre primären Endpunkte für den klinischen Nutzen nicht. Der klinische Nutzen wurde anhand einer standardisierten Skala bewertet, die den Verlauf der grobmotorischen Leistung misst, einschließlich Stehen, Gehen, Springen und Kopfheben.
Zweitens konnten alternative Endpunkte, die geringfügig günstiger waren – und die letztlich von der Food and Drug Administration (FDA) zur Unterstützung der Zulassung herangezogen wurden – nicht durch statistische Tests verifiziert werden.
Darüber hinaus war das primäre alternative Ergebnis der Spiegel des Mikrodystrophin-Proteins, ein Biomarker, der kürzlich in einer anderen DMD-Gentherapiestudie keine klinische Relevanz zeigte. [2]
Wann wurde Elevidys genehmigt?
Trotz der begrenzten klinischen Beweise erhielt Delandistrogen Moxeparvovec-Rokl am 22. Juni 2023 die FDA-Zulassung als erste Genersatztherapie für die Duchenne-Muskeldystrophie (DMD), speziell für gehfähige Kinder im Alter von 4 bis 5 Jahren. [3]
Die Zulassung wurde im Rahmen des beschleunigten Zulassungsprogramms des FDA erteilt. [4]
Am 20. Juni 2024 erweiterte die FDA ihre Zulassung um die herkömmliche Zulassung für gehfähige Personen ab 4 Jahren, bei denen die Krankheit diagnostiziert wurde und die eine bestätigte Mutation im DMD-Gen aufweisen. Die Behörde hat außerdem eine beschleunigte Zulassung für Personen ab 4 Jahren erteilt, die auf Rollstühle angewiesen sind. [5]
Im klinischen Überprüfungsdokument von FDA wurde eine besorgniserregende Feststellung bezüglich der erweiterten Zulassung gemacht: [6]
Dr. Peter Marks, Direktor des Center for Biologics Evaluation and Research (CBER), hat dem Antrag stattgegeben und dabei die Empfehlung des Gutachterteams außer Kraft gesetzt.
Delandistrogene Moxeparvovec-Rokl ist nun in den USA für den Vertrieb zugelassen. Der Preis für die Therapie wurde kürzlich auf über $3 Millionen pro Behandlung festgesetzt. [7]
Die Muskeldystrophie Duchenne (DMD) ist eine seltene, fortschreitende und tödlich verlaufende Erkrankung, für die es nur wenige wirksame Behandlungsalternativen gibt.
Mutationen der Muskeldystrophie Duchenne (DMD) sind hauptsächlich rezessiv und X-chromosomal, was dazu führt, dass sie vorwiegend Männer betreffen. Die Häufigkeit der Geburt bei Männern liegt bei etwa 1 zu 3.600. [Weiterlesen: Was ist DMD?]
Diese Mutationen führen zum Abbau der Skelett- und Herzmuskulatur, der sich bereits in der frühen Kindheit manifestiert, obwohl der Bewegungsapparat ansonsten normal entwickelt ist. Personen mit einem DMD-Genotyp verlieren typischerweise im frühen Jugendalter ihre Gehfähigkeit und haben eine Lebenserwartung, die oft erst in den Dreißigern endet.
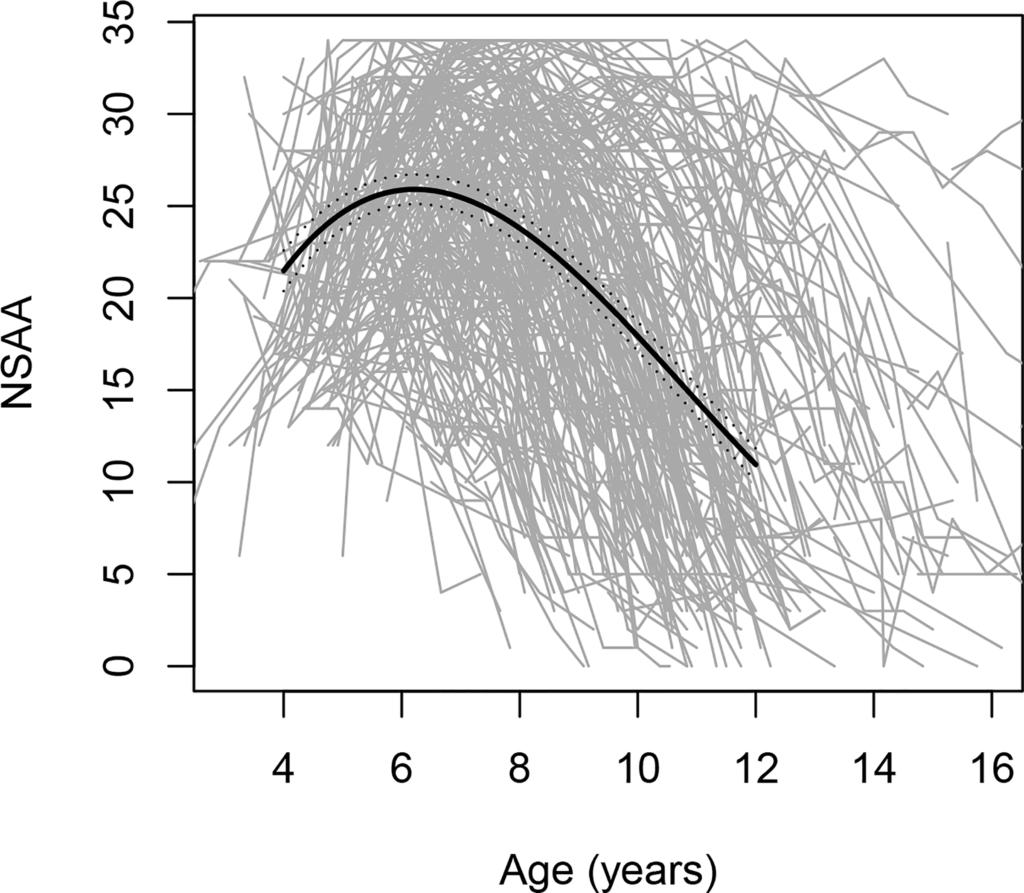
Der Krankheitsverlauf ist heterogen, wie aus FDA-Daten hervorgeht, die eine erhebliche Variabilität der motorischen Funktionstrends bei Jungen mit DMD im Alter von 4 bis 16 Jahren anzeigen (siehe Abbildung).
Figur. Veränderungen der Ergebnisse des North Star Ambulatory Assessment (NSAA), einer Skala zur Messung der grobmotorischen Leistungsfähigkeit, mit dem Alter; umfasst 395 einzelne Verläufe, die jeweils durch eine graue Linie dargestellt werden.
Sareptas DMD-Heilmittel
Delandistrogene Moxeparvovec-Rokl stellt die erste krankheitsmodifizierende Therapie für die Mehrzahl der Fälle der Duchenne-Muskeldystrophie (DMD) dar. Allerdings hatte das FDA zuvor bereits vier Medikamente für weniger verbreitete Varianten von DMD zugelassen.
Bei Eteplirsen (Exondys 51) handelt es sich um ein Medikament, das derzeit intensiv untersucht wird. Es wurde entwickelt, um bestimmte DMD-Mutationen durch den Einsatz von Antisense-Oligonukleotiden zu behandeln, die ein gezieltes „Exon-Skipping“ ermöglichen und dadurch mutierte Regionen bei der Gen-zu-Protein-Expression umgehen. [8]
Es gibt fünf FDA-zugelassene Heilmittel für DMD, darunter Delandistrogen Moxeparvovec-Rokl, die alle verkürzte Formen des essentiellen Muskelproteins Dystrophin verwenden (siehe unten). [9]
Alle diese Heilmittel wurden im Rahmen des beschleunigten Zulassungsprogramms von FDA zugelassen. Obwohl sie bereits seit mehreren Jahren bei Patienten angewendet werden, haben die meisten dieser Heilmittel keine signifikanten klinischen Vorteile gegenüber Placebos gezeigt.
Darüber hinaus werden vier der Heilmittel, darunter Delandistrogen Moxeparvovec-Rokl, von einem einzigen Unternehmen, Sarepta Therapeutics, hergestellt. [10]
Dr. Robert Califf, der FDA-Kommissar während der Zulassung von Eteplirsen und Delandistrogen Moxeparvovec-Rokl, äußerte anhaltende Bedenken hinsichtlich der DMD-Produkte von Sarepta im Jahr 2024 und erklärte: „Sarepta ist wie ein Fluch für mich.“ [11]
Wirkungsmechanismus von Elevidys
Bei Mutationen, die DMD verursachen, ist die Expression eines Gens, das für das Protein Dystrophin kodiert, deutlich reduziert oder unterdrückt.
Viele biochemische Bestandteile des Proteins passieren die Membranen der Muskelzellen. Darüber hinaus dient es als Stoßdämpfer und hält die Muskeln stark, auch wenn sie durch häufige Kontraktions-Expansions-Zyklen geschwächt werden könnten. [12]
Theoretisch kann DMD gemildert oder sogar rückgängig gemacht werden, indem die Dystrophin-Expression in den Muskelzellen der Betroffenen gefördert wird. Als Reaktion auf diese Annahme wurde das Delandistrogen Moxeparvovec-Rokl entwickelt, dessen aktive Komponente eine modifizierte RNA-Sequenz ist, die für eine verkürzte Variante von Dystrophin, das sogenannte „Mikrodystrophin“, kodiert.
Mikro-Dystrophin hat etwa 30% der Größe von nicht mutiertem Dystrophin. Das verkürzte Protein, das bei Patienten mit Muskeldystrophie vom milderen Typ Becker beobachtet wird, diente als Grundlage für die Erfindung des Mikrodystrophins, das durch das Delandistrogen Moxeparvovec-Rokl kodiert wird.
Da die RNA-Sequenz in Viruskapsiden gebündelt werden musste, um in den Körper gelangen zu können, musste das Dystrophin-Gen gekürzt werden. Die vollständige Dystrophin-RNA übersteigt die Kapazitätsgrenze solcher Kapside.
Das Delandistrogen Moxeparvovec-Rokl wurde mithilfe einer komplexen Biotechnik entwickelt, die ein Protein hervorbringt, das die Dystrophinfunktion im gesamten Körper höchstens teilweise wiederherstellen würde. Die Wirksamkeit dieser komplizierten Therapie wurde in klinischen Studien noch nicht nachgewiesen.
Beschleunigte Zulassung von 2023
An der entscheidenden Studie zur Erstzulassung von Delandistrogen Moxeparvovec-Rokl nahmen 41 gehfähige Kinder mit DMD im Alter von 4 bis 7 Jahren teil. Sie wurden im Verhältnis 1:1 randomisiert und erhielten das Medikament oder ein Placebo.
Das wichtigste vorherbestimmte klinische Ergebnis war die Veränderung der grobmotorischen Leistungsfähigkeit über einen Zeitraum von 48 Monaten, gemessen anhand einer standardisierten Skala mit 17 Punkten. Bei den mit Delandistrogen Moxeparvovec-Rokl behandelten Patienten verbesserte sich die Leistungsfähigkeit im Vergleich zur Kontrollgruppe nicht signifikant.13]
Basierend auf dem Surrogatendpunkt der Mikrodystrophinexpression im Muskelgewebe 12 Wochen nach der Gentherapie beantragte der Sponsor Sarepta aufgrund des ungünstigen Ergebnisses der klinischen Studie eine schnelle Zulassung.
Obwohl nach 12 Wochen eine Mikrodystrophin-Expression festgestellt wurde, gab es keine Korrelation zwischen den Mikrodystrophin-Werten und der daraus resultierenden Muskelfunktion. Folglich kamen die wissenschaftlichen Gutachter von FDA zu dem Schluss, dass die Delandistrogen-Moxeparvovec-Rokl-Studie keinen therapeutischen Nutzen für die Behandlung von DMD brachte. Laut der klinischen FDA-Bewertung aus dem Jahr 2023: [14]
Daher reichen diese Daten sogar für eine kleine Population, wie etwa ambulante Patienten mit DMD im Alter von 4 bis 5 Jahren, die eine bestätigte DMD-Mutation in ihrem DMD-Gen aufweisen, nicht aus, um die Expression von ELEVIDYS-Mikrodystrophin als Surrogat-Endpunkt zu unterstützen, der „mit hinreichender Wahrscheinlichkeit einen klinischen Nutzen vorhersagt“ für die beschleunigte Zulassung von ELEVIDYS.
Der Direktor des Center for Biologics Evaluation and Research von FDA ignorierte die wissenschaftlichen Gutachter der Behörde und entschied sich trotz dieser harschen Kritik für eine schnelle Zulassung für gehfähige 4- bis 5-jährige Kinder mit DMD.
Als primäre Grundlage für die Entscheidung dienten Daten von 8 der 41 Patienten. Darüber hinaus sei der Sponsor „als Voraussetzung für die beschleunigte Zulassung“ verpflichtet, eine zweite randomisierte Studie abzuschließen, deren Ergebnisse bis Herbst 2023 erwartet würden, heißt es in dem Urteil.15]
Erweiterte Zulassung im Jahr 2024
Sarepta teilte in einer Pressemitteilung vom 30. Oktober 2023 mit, dass der primäre Endpunkt der zweiten entscheidenden randomisierten Studie mit dem Delandistrogen Moxeparvovec-Rokl nicht erreicht wurde.
In dieser placebokontrollierten Studie wurden 125 Patienten im Alter zwischen 4 und 7 Jahren im Verhältnis 1:1 randomisiert entweder einem Placebo oder dem Delandistrogen Moxeparvovec-Rokl zugeteilt.16]
Der wichtigste Endpunkt war eine 52-wöchige Veränderung auf derselben Muskelfunktionsskala wie im vorherigen Experiment. Auf der standardisierten grobmotorischen Funktionsskala (mit einem maximalen Bereich von 34 Punkten) wurde zwischen dem Medikament und dem Placebo nur ein durchschnittlicher Unterschied von 0,65 Punkten festgestellt, sodass die Ergebnisse nicht signifikant waren (p=0,25).
Das FDA hätte die Zulassung von Delandistrogen Moxeparvovec-Rokl dort stoppen sollen. Bei der Zulassung im Jahr 2023 betonte der Hersteller stattdessen sekundäre Ergebnisse, die seiner Aussage nach „robuste, statistisch signifikante Ergebnisse … [die] eine Wirksamkeitsergänzung unterstützen“ erbrachten.
Die wissenschaftlichen Gutachter von FDA lehnten diese neuen Erkenntnisse scharf ab. Insbesondere die 10-Meter-Geh-/Lauftests und die Zeit, die zum Aufstehen aus einer Bodenposition benötigt wurde, waren zwei wichtige sekundäre Ergebnisse.
Die Probanden mit Delandistrogen-Moxeparvovec-Rokl führten diese Aktivitäten im Durchschnitt 0,5 Sekunden schneller aus als die Placebo-Probanden, die zu Beginn 3,5 bzw. 4,9 Sekunden benötigten.
Diese Erkenntnisse, die weder vorab festgelegt noch für die multiplen Datenanalysen statistisch korrigiert wurden, „können die Wirksamkeit von ELEVIDYS nicht belegen“, so die Gutachter von FDA.
Den Gutachtern von FDA zufolge waren die Daten zudem „irreführend und können keinem Beteiligten – einschließlich Patienten, Familienangehörigen, Pflegepersonal und verschreibenden Ärzten – dabei helfen, fundierte Entscheidungen über den möglichen Nutzen einer Behandlung mit ELEVIDYS zu treffen.“ [17]
Im Juni 2024 überstimmte der CBER-Direktor jedoch sein Team und genehmigte das Delandistrogen Moxeparvovec-Rokl zum zweiten Mal, trotz der ungünstigen Bewertungen der FDA-Wissenschaftler. [18]
Wie bereits erwähnt, wurde mit der zweiten Zulassung die Zulassung erweitert und verschärft, um eine typische Zulassung für ambulante Patienten mit der Erkrankung ab 4 Jahren und nachgewiesener Mutation im DMD-Gen einzuschließen. Darüber hinaus wurde in der zweiten Zulassung eine beschleunigte Zulassung für rollstuhlabhängige Menschen ab 4 Jahren aufgenommen. [19]
Der Ersatzhinweis auf einen Zusammenhang zwischen den Mikrodystrophin-Proteinwerten und der Zeit bis zum Aufstehen aus der Liegeposition war ein wichtiger Faktor im Entscheidungsmemorandum des CBER-Direktors für 2024.
Dieser Zusammenhang war jedoch aufgrund der geringen Steigung (-0,012 Sekunden für jede prozentuale Änderung des Mikrodystrophinspiegels im Muskel) weder optisch (grafisch) überzeugend noch statistisch signifikant (p=0,1388).
Darüber hinaus wichen zahlreiche Datenpunkte erheblich von der geschätzten Regressionslinie ab.
Da nur 25% der Teilnehmer der zweiten zentralen Studie in diese Analyse einbezogen wurden, hatten die Experten ausdrücklich darauf hingewiesen, dass die Ergebnisse bei der Bewertung der Mikrodystrophin-Dosis-Wirkungs-Daten „mit Vorsicht interpretiert“ werden sollten. Obwohl die Verwendung des Mikrodystrophin-Gens zur Behandlung von DMD wissenschaftlich gerechtfertigt ist, haben klinische Studien daher keine positiven Auswirkungen der Gentherapie gezeigt.20]
Tatsächlich wurde in einer kürzlich durchgeführten Phase-3-Studie einer von Pfizer entwickelten DMD-Gentherapie eine „signifikante Menge“ von Mikrodystrophin in den Muskeln von DMD-Patienten festgestellt, aber es kam zu keiner entsprechenden klinischen Verbesserung. Berichten zufolge hat Pfizer die Arbeit an der Heilung eingestellt. [21]
Kommentare der Familie
Ähnliche Sorgen wurden von Menschen geäußert, die direkt von DMD betroffen sind. Bedenken hinsichtlich der Sicherheit und Wirksamkeit von Delandistrogen Moxeparvovec-Rokl veranlassten einen Elternteil im September 2024 zu schreiben, dass er sich entschieden habe, bei seinem Kind keine Therapie zu machen. [22]
In einem Online-Video kritisierte ein anderer Elternteil Sarepta für die Entwicklung eines Heilmittels, das unzureichend sei und bald veraltet sein werde. Als Reaktion darauf wurde einer Eltern-Selbsthilfegruppe, die das Video gehostet hatte, vorgeworfen, die Videokritik von ihrer Website entfernt zu haben, nachdem Sarepta Therapeutics gedroht hatte, die Gruppe nicht mehr zu unterstützen.23]
Lohnen sich die Kosten für Elevidys im Vergleich zu klinischen Studien?
Ist der Preis von 3 Millionen US-Dollar für die Elevidys-Gentherapie angesichts der Ergebnisse der klinischen Studien angemessen? [Weiterlesen: Muskeldystrophie Duchenne: Behandlung und Kosten]
Wenn man die Menge an Dystrophin berücksichtigt, die nach der Anwendung von Elevidys im Körper produziert wird, und die in klinischen Studien ermittelten Ergebnisse des North Star Ambulatory Assessment (NSAA) berücksichtigt, sind 3 Millionen USD für diese Behandlungsmethode sehr viel.
Unser Aufruf an Sarepta, den Entwickler von Elevidys, und an Roche, die globale Marketingbehörde: Reduzieren Sie diesen für diesen gewerblichen Zweck festgelegten hohen Preis sofort!
Mehr erfahren: Mögliche neue Gentherapien für die Muskeldystrophie Duchenne
Erste Todesanzeige
Am 18. März 2025 gab Sarepta Therapeutics bekannt, dass der erste registrierte Todesfall im Zusammenhang mit ihrer Gentherapie für die Muskeldystrophie Duchenne, Elevidys, bei einem 16-jährigen Jungen aufgetreten sei. [Mehr lesen]
Abschluss
Ein Zentrumsleiter setzte sich gegen das wissenschaftliche Team von FDA durch, das mit der Beurteilung des Medikaments betraut war, und FDA genehmigte Delandistrogen Moxeparvovec-Rokl als Gentherapie für DMD.
Die beiden zentralen Studien zu Delandistrogene Moxeparvovec-Rokl haben ihre wichtigsten klinischen Ziele nicht erreicht. Indikatoren, die zur Rettung der Anwendung von Delandistrogene Moxeparvovec-Rokl verwendet wurden, wie sekundäre Endpunkte (z. B. 10-Meter-Geh-/Lauftests) und der indirekte Surrogatindikator für Mikrodystrophinwerte, waren an und für sich problematisch.
Aus diesem Grund sind wir der Meinung, dass die Führung von FDA einen riskanten Präzedenzfall geschaffen hat, der die Öffentlichkeit nicht davor schützt, einem teuren und körperlich anstrengenden, unwirksamen Medikament ausgesetzt zu werden. [24]
Mehr erfahren: Markt für Muskeldystrophie Duchenne wächst: Doch nicht alle Familien haben Zugang zu Behandlungen
Eine Möglichkeit, auf das Urteil FDA zu reagieren, besteht darin, zu sagen, dass schwache Zulassungskriterien das Problem seien und dass strengere Zulassungsstandards und Verfahren nach der Markteinführung die Lösung seien. [25]
Die internen Prozesse von FDA sollten geändert werden, um es einzelnen Beamten in Führungspositionen deutlich zu erschweren, die Konsensbewertung der mit einer Regulierungsentscheidung betrauten Wissenschaftler der Behörde zu überstimmen, auch wenn wir der Meinung sind, dass strengere Zulassungsstandards und Anforderungen nach der Markteinführung erforderlich sind.
Laut Califf, der zum Zeitpunkt der beiden Zulassungsentscheidungen für Delandistrogen Moxeparvovec-Rokl als Commissioner für FDA zuständig war, haben die Gegner der Entscheidung eine „sehr vereinfachte Sicht auf klinische Beweise“ hinsichtlich der Schwierigkeiten bei der Entwicklung von Therapien für schwere und seltene Erkrankungen. [26]
Wir stimmen dieser Einschätzung nicht zu. Das FDA sollte seinen Entscheidungsprozess für neue Arzneimittel, egal ob für häufige oder seltene Krankheiten, aufgrund der Zulassung von Delandistrogen Moxeparvovec-Rokl verbessern.
Mehr erfahren: Türkischer Vertreter von DMD WarrioR teilt seine Ansichten zur Elevidys-Gentherapie
In einem Interview vor seinem Ausscheiden sprach Califf auch über die Entscheidung und wies darauf hin, dass Familien mit solch schweren und seltenen Krankheiten verständlicherweise nach Hoffnung dürsten und dass er als politischer Kandidat auch nicht bereit sei, die Entscheidungen der hauptberuflichen FDA-Mitarbeiter aufzuheben. Diese Faktoren sind zwar wichtig, reichen jedoch nicht aus, um die Entscheidung des FDA zu stützen, das Delandistrogen Moxeparvovec-Rokl zuzulassen. [27]

{kind=link}