
Quelle est l'efficacité de la thérapie génique ELEVIDYS (delandistrogene moxeparvovec-rokl), qui a été introduite sur le marché pharmaceutique, par rapport à son coût de 3 millions de dollars américains ? L'examen des essais cliniques de la thérapie génique Elevidys sera le sujet principal de cet essai. Nous évaluerons le coût et l'efficacité de cette thérapie génique, qui a été développée pour traiter la dystrophie musculaire de Duchenne. Le coût du Elevidys est-il rentable par rapport aux essais cliniques ?
Table des matières
Aperçu des essais cliniques
La Food and Drug Administration (FDA) a autorisé une thérapie génique révolutionnaire en 2023 et 2024 pour la dystrophie musculaire de Duchenne, une maladie rare mais mortelle débutant dans l'enfance.
Seuls les jeunes âgés de 4 à 5 ans capables de marcher (déambulatoires) étaient éligibles à l'autorisation 2023.
La décision de 2024 a élargi cette autorisation pour couvrir l’approbation conventionnelle des patients ambulatoires atteints de la maladie et d’une mutation vérifiée dans le gène DMD qui sont âgés de 4 ans ou plus.
De plus, le FDA a établi une approbation accélérée pour les personnes de 4 ans et plus qui dépendent de fauteuils roulants.
Aucune de ces approbations n’a été appuyée par des résultats d’essais cliniques appropriés montrant que les enfants atteints de DMD conservaient ou rétablissaient leur capacité motrice globale, ni par des évaluations scientifiques positives du FDA.
Un directeur du FDA a ignoré le personnel scientifique de l'agence, ce qui a donné lieu aux deux approbations.
Cet article analyse les deux approbations mal réfléchies ainsi que les raisons pour lesquelles les dirigeants du FDA ne devraient pas suivre leur exemple à l'avenir. Il propose également des suggestions sur la manière dont le FDA peut modifier la manière dont il gère les décisions réglementaires controversées.
Comment Elevidys a été approuvé par FDA
Les deux essais cliniques randomisés de phase 3 du delandistrogene moxeparvovec-rokl (Elevidys) comme traitement de remplacement génique pour la dystrophie musculaire de Duchenne (DMD) ont été rapportés dans Nature Medicine le 29 octobre 2024. [1] L’efficacité clinique de ce médicament n’a pas été prouvée de manière concluante pour deux raisons, selon ce rapport.
Les deux essais n’ont pas atteint leurs principaux critères d’évaluation en termes de bénéfice clinique. Le bénéfice clinique a été évalué à l’aide d’une échelle standardisée qui mesure la trajectoire des performances motrices globales, notamment la station debout, la marche, le saut et le soulèvement de la tête.
Deuxièmement, les critères d’évaluation alternatifs qui étaient marginalement favorables – finalement utilisés par la Food and Drug Administration (FDA) pour justifier l’approbation – n’ont pas pu être vérifiés par des tests statistiques.
De plus, le principal résultat alternatif était les niveaux de protéine micro-dystrophine, un biomarqueur qui n'a récemment pas montré de pertinence clinique dans un autre essai de thérapie génique DMD. [2]
Quand le Elevidys a-t-il été approuvé ?
Malgré les preuves cliniques limitées, le 22 juin 2023, le delandistrogene moxeparvovec-rokl a reçu l'approbation FDA comme première thérapie de remplacement génique pour la dystrophie musculaire de Duchenne (DMD), spécifiquement pour les enfants ambulatoires âgés de 4 à 5 ans. [3]
L'approbation a été obtenue grâce au programme d'approbation accéléré du FDA. [4]
Le 20 juin 2024, le FDA a élargi son approbation pour inclure l'autorisation traditionnelle pour les personnes ambulatoires âgées de 4 ans et plus diagnostiquées avec la maladie et possédant une mutation confirmée du gène DMD. L'agence a également accordé une approbation accélérée pour les personnes âgées de 4 ans et plus qui dépendent d'un fauteuil roulant. [5]
Une déclaration préoccupante a été notée dans le document d'examen clinique du FDA concernant l'action d'approbation élargie : [6]
Le Dr Peter Marks, directeur du Centre d'évaluation et de recherche sur les produits biologiques (CBER), a approuvé la demande en annulant la recommandation de l'équipe d'évaluation.
Le delandistrogene moxeparvovec-rokl est désormais autorisé à la commercialisation aux États-Unis. Le prix de la thérapie a récemment été fixé à plus de 143 millions de dollars par traitement. [7]
La dystrophie musculaire de Duchenne (DMD) est une maladie rare, progressive et mortelle caractérisée par une pénurie d’alternatives thérapeutiques efficaces.
Les mutations de la dystrophie musculaire de Duchenne (DMD) sont principalement récessives et liées au chromosome X, ce qui entraîne un impact prédominant sur les hommes. L'incidence de la naissance chez les hommes est d'environ 1 sur 3 600. [Lire la suite : Qu'est-ce que la DMD ?]
Ces mutations entraînent une dégradation des muscles squelettiques et cardiaques, qui se manifeste dès la petite enfance, malgré un développement musculo-squelettique par ailleurs normal. Les personnes atteintes d'un génotype DMD perdent généralement la capacité de marcher au début de l'adolescence et ont une espérance de vie qui se termine souvent vers la trentaine.
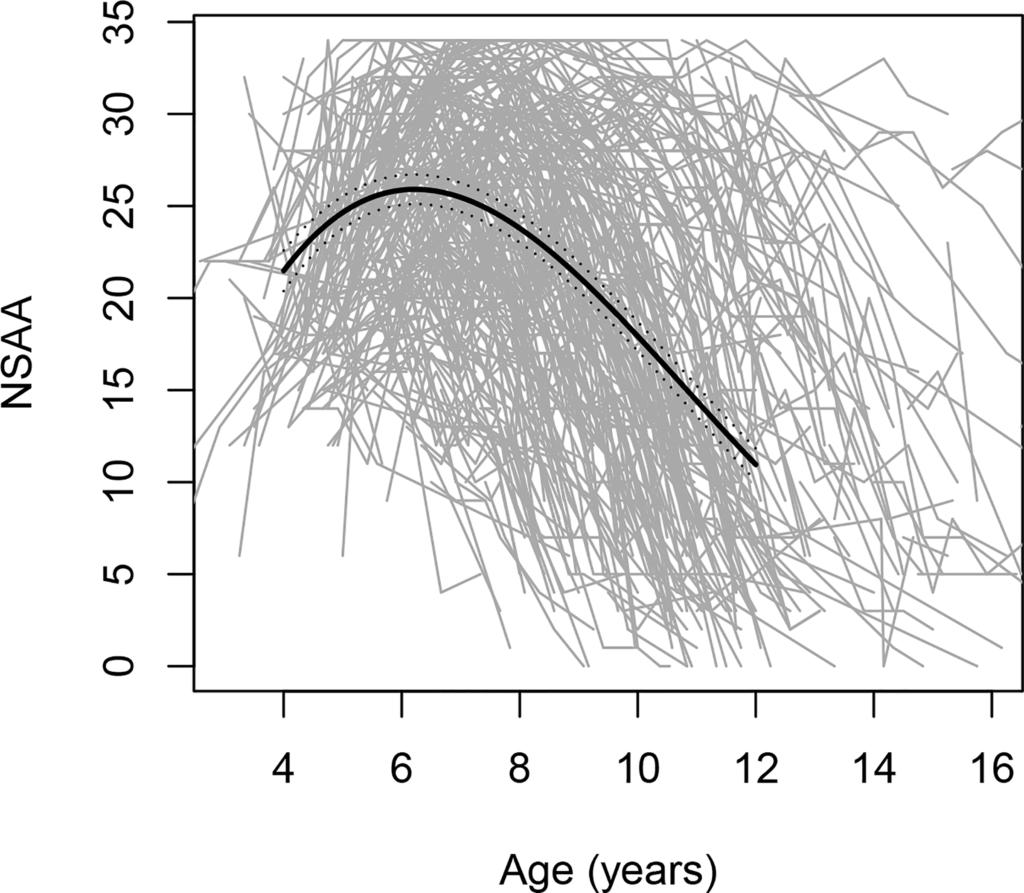
L'évolution de la maladie est hétérogène, comme en témoignent les données FDA indiquant une variabilité significative des tendances de la fonction motrice chez les garçons atteints de DMD âgés de 4 à 16 ans (voir la figure).
Chiffre. Évolution des scores de l'évaluation ambulatoire North Star (NSAA), une échelle de performance motrice globale, avec l'âge ; comprend 395 trajectoires individuelles, chacune représentée par une ligne grise.
Les remèdes contre la DMD de Sarepta
Le delandistrogene moxeparvovec-rokl représente le traitement modificateur de la maladie inaugural pour la majorité des cas de dystrophie musculaire de Duchenne (DMD) ; cependant, le FDA avait précédemment approuvé quatre médicaments pour des variantes moins courantes de DMD.
Eteplirsen (Exondys 51) est un médicament à l'étude, conçu pour traiter des mutations DMD spécifiques grâce à l'utilisation d'oligonucléotides antisens qui permettent le « saut d'exon » ciblé, contournant ainsi les régions mutées dans l'expression gène-protéine. [8]
Il existe cinq traitements approuvés par le FDA pour la DMD, y compris le delandistrogene moxeparvovec-rokl, qui utilisent tous des formes tronquées de la protéine musculaire essentielle dystrophine (comme détaillé ci-dessous). [9]
Tous ces traitements ont été approuvés via le programme d'approbation accélérée FDA. Bien qu'ils soient utilisés chez les patients depuis plusieurs années, la majorité de ces traitements n'ont pas montré d'avantages cliniques significatifs par rapport au placebo.
De plus, quatre des remèdes, dont le delandistrogene moxeparvovec-rokl, sont produits par une seule entreprise, Sarepta Therapeutics. [10]
Le Dr Robert Califf, commissaire du FDA lors de l'approbation de l'eteplirsen et du delandistrogene moxeparvovec-rokl, a exprimé ses inquiétudes persistantes concernant les produits DMD de Sarepta en 2024, déclarant : « Sarepta est comme une malédiction pour moi. » [11]
Mécanisme d'action du Elevidys
L’expression d’un gène codant pour la protéine dystrophine est considérablement réduite ou réduite au silence dans les mutations qui causent la DMD.
De nombreux composants biochimiques de la protéine traversent les membranes des cellules musculaires. De plus, elle sert d'amortisseur, préservant la force des muscles face à la détérioration potentielle provoquée par des cycles fréquents de contraction-expansion. [12]
En théorie, la DMD peut être atténuée, voire inversée, en favorisant l’expression de la dystrophine dans les cellules musculaires des personnes atteintes. Le delandistrogene moxeparvovec-rokl, dont le composant actif est une séquence d’ARN modifiée qui code pour une variante raccourcie de la dystrophine connue sous le nom de « micro-dystrophine », a été développé en réponse à cette idée.
La microdystrophine a une taille d'environ 30% de celle de la dystrophine non mutée. La protéine raccourcie observée chez les patients atteints du type plus léger de dystrophie musculaire de Becker a servi de base à l'invention de la micro-dystrophine codée par le delandistrogène moxeparvovec-rokl.
Comme la séquence d'ARN doit être regroupée dans des capsides virales pour être délivrée dans l'organisme, le gène de la dystrophine a dû être tronqué. L'ARN de la dystrophine complet dépasse la limite de capacité de ces capsides.
Le delandistrogene moxeparvovec-rokl a été créé à l'aide d'une technique de bio-ingénierie complexe qui a permis d'obtenir une protéine qui, au mieux, ne restaurerait que partiellement la fonction de la dystrophine dans tout l'organisme. L'efficacité de cette thérapie complexe n'a pas encore été démontrée dans les essais cliniques.
Approbation accélérée de 2023
Quarante et un enfants ambulatoires atteints de DMD, âgés de 4 à 7 ans, ont participé à l'essai pivot pour la première approbation du delandistrogene moxeparvovec-rokl. Ils ont été randomisés 1:1 pour recevoir le médicament ou un placebo.
Le principal résultat clinique prédéterminé était le changement de la performance motrice globale sur une période de 48 mois telle que mesurée par une échelle standardisée de 17 éléments ; les patients traités avec du delandistrogene moxeparvovec-rokl n'ont pas amélioré significativement leurs performances par rapport au groupe témoin. [13]
Sur la base du critère de substitution de l'expression de la micro-dystrophine dans le tissu musculaire 12 semaines après la thérapie génique, Sarepta, le sponsor, a demandé une approbation rapide en raison du résultat défavorable de l'essai clinique.
Malgré le fait que l'expression de la microdystrophine ait été notée à 12 semaines, il n'y avait aucune corrélation entre les niveaux de microdystrophine et la fonction musculaire qui en résultait. Par conséquent, les examinateurs scientifiques de FDA sont arrivés à la conclusion qu'il n'y avait aucun bénéfice thérapeutique à traiter la DMD à partir de l'essai delandistrogene moxeparvovec-rokl. Selon l'examen clinique FDA de 2023 : [14]
Par conséquent, même pour une petite population, comme les patients ambulatoires atteints de DMD âgés de 4 à 5 ans qui ont une mutation DMD confirmée dans leur gène DMD, ces données sont insuffisantes pour soutenir l'expression de la micro-dystrophine ELEVIDYS comme critère d'évaluation de substitution « raisonnablement susceptible de prédire un bénéfice clinique » pour l'approbation accélérée d'ELEVIDYS.
Le directeur du Centre d'évaluation et de recherche des produits biologiques du FDA a ignoré les évaluateurs scientifiques de l'agence et a décidé d'accorder une approbation rapide aux enfants ambulatoires de 4 et 5 ans atteints de DMD malgré ces critiques sévères.
Les données de 8 des 41 patients ont servi de base principale à la décision. En outre, le promoteur était « tenu, comme condition d’approbation accélérée », de terminer un deuxième essai randomisé, dont les résultats étaient attendus d’ici l’automne 2023, selon la décision.15]
Approbation élargie en 2024
Sarepta a déclaré dans un communiqué de presse daté du 30 octobre 2023 que le critère d'évaluation principal du deuxième essai pivot randomisé du delandistrogene moxeparvovec-rokl n'avait pas été atteint.
Dans cette étude contrôlée par placebo, 125 patients âgés de 4 à 7 ans ont été répartis aléatoirement 1:1 pour recevoir soit un placebo, soit le delandistrogene moxeparvovec-rokl. [16]
Le critère d'évaluation principal était l'évolution sur 52 semaines du score de la même échelle de fonction musculaire utilisée dans l'expérience précédente. Seule une différence moyenne de 0,65 point entre le médicament et le placebo sur l'échelle fonctionnelle motrice brute standardisée (qui a une plage maximale de 34 points) a été constatée, ce qui rend les résultats non significatifs (p = 0,25).
Le FDA aurait dû arrêter d’approuver le delandistrogene moxeparvovec-rokl à ce stade. Pour l’approbation de 2023, le fabricant a plutôt mis l’accent sur les résultats secondaires qui, selon lui, ont produit « des résultats robustes et statistiquement significatifs… [qui] soutiennent un supplément d’efficacité ».
Les évaluateurs scientifiques de l'étude FDA ont sévèrement rejeté ces nouvelles conclusions. En particulier, les tests de marche/course de 10 mètres et le temps nécessaire pour se relever d'une position au sol étaient deux résultats secondaires importants.
Les sujets Delandistrogene moxeparvovec-rokl ont réalisé ces activités en moyenne 0,5 seconde plus rapidement que les sujets placebo, qui ont mis respectivement 3,5 et 4,9 secondes au départ.
Ces résultats, qui n’ont été ni préétablis ni corrigés statistiquement pour les multiples analyses de données, « ne peuvent pas soutenir l’efficacité d’ELEVIDYS », selon les évaluateurs du FDA.
Les données, selon les évaluateurs du FDA, étaient également « trompeuses et ne peuvent guider aucune partie prenante — y compris les patients, les membres de la famille et les soignants, ainsi que les prescripteurs — dans la prise de décisions éclairées sur les avantages potentiels de la guérison avec ELEVIDYS. » [17]
Cependant, en juin 2024, le directeur du CBER a annulé la décision de son équipe et approuvé le delandistrogene moxeparvovec-rokl pour la deuxième fois, malgré les critiques défavorables des scientifiques du FDA. [18]
Comme mentionné précédemment, la deuxième approbation a élargi et renforcé l'autorisation pour inclure l'approbation typique chez les patients ambulatoires atteints de la maladie qui sont âgés de 4 ans ou plus et qui ont une mutation vérifiée dans le gène DMD. De plus, une autorisation accélérée pour les personnes dépendantes d'un fauteuil roulant âgées de 4 ans et plus a été incorporée dans la deuxième approbation. [19]
L'indication de substitution du lien entre les niveaux de protéines de micro-dystrophine et le temps nécessaire pour se lever après être resté allongé sur le sol a été un facteur majeur dans la note de décision 2024 du directeur du CBER.
Ce lien n'était cependant ni visuellement (graphiquement) convaincant ni statistiquement significatif (p = 0,1388) en raison de la faible pente (-0,012 seconde pour chaque pourcentage de changement du niveau de micro-dystrophine du muscle).
En outre, un grand nombre de points de données s’écartaient considérablement de la ligne de régression estimée.
Étant donné que seulement 25% des individus du deuxième essai pivot ont été inclus dans cette analyse, les experts de FDA ont explicitement noté que les résultats devaient être « interprétés avec prudence » lors de l’évaluation des données de dose-réponse de la micro-dystrophine. Par conséquent, malgré la justification scientifique de l’utilisation de l’insertion du gène de la micro-dystrophine pour traiter la DMD, les essais cliniques n’ont pas montré que la thérapie génique avait des effets positifs. [20]
En effet, des « quantités significatives » d’expression de micro-dystrophine ont été observées dans les muscles des patients atteints de DMD lors d’un récent essai de phase 3 d’une thérapie génique DMD développée par Pfizer, mais aucune amélioration clinique proportionnelle n’a été observée. Selon certaines informations, Pfizer a cessé de travailler sur son traitement. [21]
Commentaires de la famille
Des inquiétudes similaires ont été exprimées par les personnes directement touchées par la DMD. Les inquiétudes concernant la sécurité et l'efficacité du delandistrogene moxeparvovec-rokl ont conduit un parent à écrire en septembre 2024 qu'il avait choisi de ne pas poursuivre la thérapie pour son enfant. [22]
Dans une vidéo en ligne, un autre parent a critiqué Sarepta pour avoir créé un remède qui était insuffisant et qui deviendrait bientôt obsolète. En réponse, un groupe de soutien aux parents qui hébergeait la vidéo a été accusé d'avoir supprimé la vidéo critique de delandistrogene moxeparvovec-rokl de son site Web après que Sarepta Therapeutics a menacé de cesser de financer le groupe. [23]
Le coût du Elevidys est-il rentable par rapport aux essais cliniques ?
Compte tenu des résultats des essais cliniques, le prix de 3 millions de dollars pour la thérapie génique Elevidys est-il raisonnable ? [Lire la suite : Dystrophie musculaire de Duchenne : traitement et coût]
Compte tenu de la quantité de dystrophine produite dans le corps après l'utilisation de Elevidys et des scores d'évaluation ambulatoire North Star (NSAA) examinés dans les essais cliniques, 3 millions USD sont très exorbitants pour cette méthode de traitement.
Notre appel à Sarepta, le développeur de Elevidys, et à Roche, l'autorité mondiale en matière de marketing : Réduisez immédiatement ce prix élevé fixé à cet effet commercial !
Apprendre encore plus: Nouvelles thérapies géniques potentielles à venir pour la dystrophie musculaire de Duchenne
Première annonce de décès
Le 18 mars 2025, Sarepta Therapeutics a déclaré que le premier décès enregistré associé à leur thérapie génique pour la dystrophie musculaire de Duchenne, Elevidys, était survenu chez un garçon de 16 ans. [En savoir plus]
Conclusion
Un directeur de centre a annulé l'avis de l'équipe scientifique du FDA, qui était chargée d'évaluer le médicament, et le FDA a approuvé le delandistrogene moxeparvovec-rokl comme thérapie génique pour la DMD.
Les deux essais pivots du delandistrogene moxeparvovec-rokl n'ont pas atteint leurs principaux objectifs cliniques. Les indicateurs utilisés pour sauver l'application du delandistrogene moxeparvovec-rokl, tels que les critères d'évaluation secondaires (par exemple, les tests de marche/course de 10 mètres) et l'indicateur de substitution indirect des niveaux de micro-dystrophine, étaient problématiques en eux-mêmes.
En conséquence, nous pensons que les dirigeants du FDA ont créé un précédent risqué qui ne protège pas le public d’une exposition à un médicament inefficace, coûteux et physiquement exigeant. [24]
Apprendre encore plus: Le marché de la dystrophie musculaire de Duchenne se développe : mais toutes les familles n'ont pas accès aux traitements
Une façon de réagir à la décision du FDA est de dire que les critères d'approbation faibles sont le problème et que des normes d'approbation et des procédures post-commercialisation plus strictes sont la solution.25]
Les processus internes du FDA devraient être modifiés pour qu'il soit beaucoup plus difficile pour les responsables individuels occupant des postes de direction de passer outre l'évaluation consensuelle des scientifiques de l'agence affectés à une décision réglementaire, même si nous convenons que des normes d'approbation et des exigences post-commercialisation plus strictes sont nécessaires.
Selon Califf, le commissaire du FDA à l’époque des deux décisions d’approbation du delandistrogene moxeparvovec-rokl, ceux qui s’opposent à la décision ont une « vision très simpliste des preuves cliniques » concernant les difficultés de développement de thérapies pour les troubles graves et rares. [26]
Nous ne sommes pas d'accord avec cette évaluation. Le FDA devrait améliorer son processus de prise de décision pour les nouveaux produits pharmaceutiques, qu'ils soient destinés à des maladies courantes ou rares, à la suite de l'approbation du delandistrogene moxeparvovec-rokl.
Apprendre encore plus: Le représentant turc de DMD WarrioR partage son point de vue sur la thérapie génique Elevidys
Dans une interview avant son départ, Califf a également évoqué la décision, soulignant que les familles atteintes de maladies aussi graves et rares aspiraient naturellement à un quelconque espoir et qu'en tant que personne nommée politiquement, il était également réticent à annuler les décisions prises par les employés de carrière du FDA. Bien que ces facteurs soient importants, ils ne suffisent pas à soutenir la décision du FDA d'approuver le delandistrogene moxeparvovec-rokl. [27]

{kind=link}