
ELEVIDYS(delandistrogene moxeparvovec-rokl)基因疗法已进入医药市场,相对于其 300 万美元的成本,其效果如何?Elevidys 基因疗法的临床试验评论将成为本文的主要主题。我们将评估这种用于治疗杜氏肌营养不良症的基因疗法的成本和效果。 与临床试验相比,Elevidys 的成本是否值得?
目录
临床试验概述
美国食品药品监督管理局 (FDA) 于 2023 年和 2024 年批准了一种革命性的基因疗法,用于治疗罕见但致命的儿童期杜氏肌营养不良症。

只有年龄 4-5 岁且能够行走的青少年才有资格获得 2023 年的许可。
2024 年的裁决扩大了该批准范围,以涵盖对患有该病且已确认 DMD 基因突变且年龄在 4 岁或以上的门诊患者的常规批准。
此外,FDA 还为 4 岁及以上依赖轮椅的人士建立了加速批准。
这两项批准均没有得到适当的临床试验结果的支持,这些结果表明患有 DMD 的儿童保留或恢复了粗大运动能力,也没有得到积极的 FDA 科学评估的支持。
FDA 的一名主任无视该机构科研人员的意见,导致两项计划均获得批准。
本文讨论了这两项考虑不周的批准,并指出了 FDA 领导人今后不应再效仿的理由。本文还就 FDA 如何改变其处理有争议的监管决策的方式提出了建议。
Elevidys 如何获得 FDA 的认可
2024年10月29日,《自然医学》杂志报道了delandistrogene moxeparvovec-rokl (Elevidys)作为杜氏肌营养不良症(DMD)基因替代疗法的两项3期随机临床试验。[1] 报道称,由于两个原因,这种药物的临床疗效尚未得到最终证实。
两项试验均未达到临床益处的主要终点。临床益处通过标准化量表进行评估,该量表测量粗大运动表现的轨迹,包括站立、行走、跳跃和抬头。
其次,美国食品药品管理局(FDA)最终采用这些略微有利的替代终点来支持批准,但无法通过统计检验进行验证。
此外,主要的替代结果是微肌营养不良蛋白的水平,微肌营养不良蛋白是一种生物标志物,最近在另一项 DMD 基因治疗试验中未显示出临床相关性。[2]
Elevidys 何时获得批准?
尽管临床证据有限,2023 年 6 月 22 日,delandistrogene moxeparvovec-rokl 获得 FDA 批准,成为首个治疗杜氏肌营养不良症 (DMD) 的基因替代疗法,专门针对 4 至 5 岁的可走动儿童。[3]
该批准是通过FDA的加速批准计划获得的。[4]
2024 年 6 月 20 日,FDA 扩大了其批准范围,涵盖了对 4 岁及以上被诊断患有该疾病且 DMD 基因已确认突变的门诊患者的传统授权。该机构还对 4 岁及以上依赖轮椅的患者给予了加速批准。[5]
FDA 的临床审查文件中提到了与扩大批准行动有关的声明:[6]
生物制品评估与研究中心(CBER)主任Peter Marks博士否决了审查小组的建议,批准了该申请。
Delandistrogene moxeparvovec-rokl 现已获准在美国上市。该疗法最近定价为每例治疗费用超过 $3 百万美元。[7]
杜氏肌营养不良症 (DMD) 是一种罕见、渐进且致命的疾病,其特征是缺乏有效的治疗方法。
杜氏肌营养不良症 (DMD) 突变主要是隐性和 X 连锁的,对男性的影响最大。男性的发病率约为 1/3,600。[阅读更多: 什么是 DMD?]
这些突变会导致骨骼肌和心肌退化,尽管肌肉骨骼发育正常,但这种退化在儿童早期就已显现。患有 DMD 基因型的人通常在青春期早期就丧失了行走能力,预期寿命通常在 30 多岁时结束。
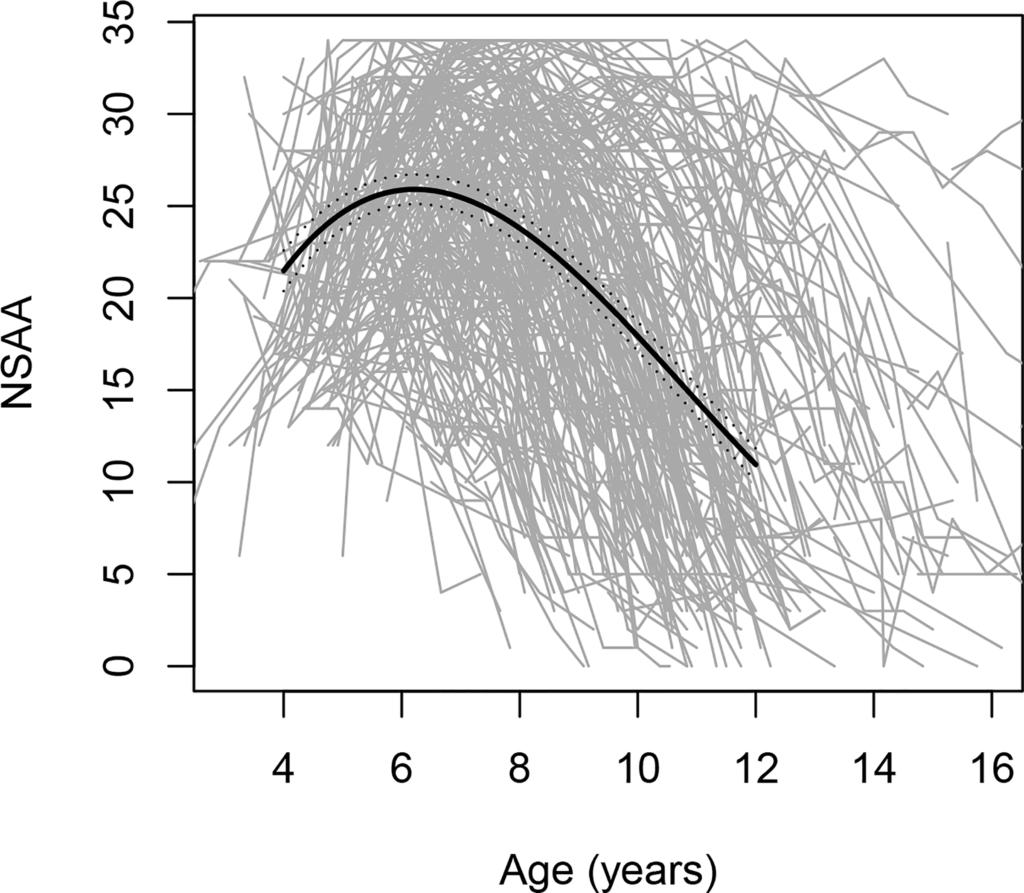
疾病过程具有异质性,FDA 数据表明,4-16 岁患有 DMD 的男孩的运动功能趋势存在显著差异(参见图).
数字。 北极星门诊评估 (NSAA) 评分(一种粗大运动表现量表)随年龄的变化;包括 395 条个人轨迹,每条轨迹都用一条灰线表示。
Sarepta 的 DMD 疗法
Delandistrogene moxeparvovec-rokl 代表了针对大多数杜氏肌营养不良症 (DMD) 病例的首个疾病改良疗法;然而,FDA 此前已批准了四种针对不太常见的 DMD 变体的药物。
Eteplirsen (Exondys 51) 是一种正在接受审查的药物,旨在通过使用反义寡核苷酸来解决特定的 DMD 突变问题,从而实现有针对性的“外显子跳跃”,从而绕过基因到蛋白质表达中的突变区域。[8]
有五种 FDA 批准的 DMD 治疗方法,包括 delandistrogene moxeparvovec-rokl,所有这些方法都利用了必需肌肉蛋白肌营养不良蛋白的截短形式(如下所述)。[9]
所有这些疗法均通过 FDA 加速审批程序获得批准。尽管这些疗法已在患者身上应用多年,但大多数疗法并未显示出比安慰剂更显著的临床优势。
此外,包括 delandistrogene moxeparvovec-rokl 在内的四种药物均由同一家公司 Sarepta Therapeutics 生产。[10]
罗伯特·卡利夫 (Robert Califf) 博士是 eteplirsen 和 delandistrogene moxeparvovec-rokl 获批期间的 FDA 专员,他对 Sarepta 2024 年的 DMD 产品表示了持续的担忧,他说:“Sarepta 对我来说就像一个诅咒。”[11]
Elevidys的作用机制
在导致 DMD 的突变中,编码蛋白质肌营养不良蛋白的基因表达显著减少或沉默。
这种蛋白质的许多生化成分都会穿过肌肉细胞膜。此外,它还充当减震器,在频繁收缩-扩张循环可能带来的肌肉退化面前保持肌肉强健。[12]
理论上,通过促进患者肌肉细胞中肌营养不良蛋白的表达,可以减轻甚至逆转 DMD 的病情。delandistrogene moxeparvovec-rokl 就是针对这一观点而开发的,其活性成分是一种经过修饰的 RNA 序列,该序列编码一种缩短的肌营养不良蛋白变体,称为“微肌营养不良蛋白”。
微肌营养不良蛋白的大小约为未突变肌营养不良蛋白的 30%。 在患有较轻的贝克尔型肌营养不良症的患者身上看到的缩短蛋白质成为发明去地那非基因 moxeparvovec-rokl 编码的微肌营养不良蛋白的基础。
由于 RNA 序列必须捆绑到病毒衣壳中才能被运送到体内,因此必须截断肌营养不良蛋白基因。完整的肌营养不良蛋白 RNA 超出了此类衣壳的容量限制。
地那非雌激素 moxeparvovec-rokl 是使用复杂的生物工程技术制造的,产生的蛋白质最多只能部分恢复全身肌营养不良蛋白的功能。这种复杂疗法的有效性尚未在临床试验中得到证实。
加速批准 2023 年
41 名 4 至 7 岁的 DMD 儿童参加了 delandistrogene moxeparvovec-rokl 首次获批的关键试验。他们按 1:1 的比例随机分配接受药物或安慰剂。
主要预定的临床结果是 48 个月内粗大运动表现的变化,以 17 项标准化量表衡量;与对照组相比,用 delandistrogene moxeparvovec-rokl 治疗的患者的表现并没有显著提高。[13]
基于基因治疗治愈后12周肌肉组织中微肌营养不良蛋白表达的替代终点,申办方Sarepta因临床试验结果不佳而请求快速批准。
尽管在 12 周时观察到微肌营养不良蛋白表达,但微肌营养不良蛋白水平与随后的肌肉功能之间没有相关性。因此,FDA 科学审阅者得出结论,delandistrogene moxeparvovec-rokl 试验对治疗 DMD 没有治疗益处。根据 2023 年的 FDA 临床审查:[14]
因此,即使对于一小部分人群,例如 4 至 5 岁患有 DMD 且 DMD 基因中已确认存在 DMD 突变的门诊患者,这些数据也不足以支持 ELEVIDYS 微肌营养不良蛋白的表达作为“合理预测临床益处”的替代终点,以加速批准 ELEVIDYS。
FDA 生物制品评估与研究中心主任不顾该机构科学评审员的意见,不顾严厉批评,决定快速批准用于 4-5 岁 DMD 儿童治疗。
41 名患者中的 8 名患者提供的数据是该决定的主要依据。此外,根据裁决,申办方“必须作为加速批准的条件”完成第二项随机试验,预计该试验的结果将于 2023 年秋季公布。[15]
2024 年扩大批准
Sarepta 在 2023 年 10 月 30 日的新闻稿中表示,地那非(delandistrogene moxeparvovec-rokl)的第二项关键随机试验的主要终点未达到。
在该安慰剂对照研究中,125 名年龄在 4 岁至 7 岁之间的患者按 1:1 的比例随机分配接受安慰剂或 delandistrogene moxeparvovec-rokl 治疗。[16]
主要终点是前次试验中所使用的相同肌肉功能量表评分在 52 周内的变化。在标准化粗大运动功能量表(最大范围为 34 分)上,药物和安慰剂之间仅发现 0.65 分的平均差异,因此结果并不显著(p=0.25)。
FDA 应该就此停止批准 delandistrogene moxeparvovec-rokl。对于 2023 年的批准,制造商反而强调了次要结果,他们说这些结果产生了“稳健、具有统计学意义的结果……[这] 支持功效补充”。
FDA 科学评审员严厉驳斥了这一新发现。特别是,10 米步行/跑步测试和从地面站起所需的时间是两个重要的次要结果。
Delandistrogene moxeparvovec-rokl 受试者完成这些活动的时间平均比安慰剂受试者快 0.5 秒,后者在基线时分别花费 3.5 秒和 4.9 秒。
FDA 审阅人员表示,这些结果既不是预先设定的,也没有针对多项数据分析进行统计校正,“无法支持 ELEVIDYS 的有效性”。
FDA 审查人员认为,这些数据也“具有误导性,无法指导任何利益相关者(包括患者、家属、护理人员和处方人员)就使用 ELEVIDYS 治愈的潜在益处做出明智的决定。”[17]
然而,2024 年 6 月,CBER 主任推翻了他的团队的决定,尽管 FDA 科学家的评价不佳,但还是第二次批准了 delandistrogene moxeparvovec-rokl。[18]
如前所述,第二次批准扩大并加强了许可范围,包括对 4 岁及以上且已证实 DMD 基因突变的门诊患者进行典型批准。此外,第二次批准还纳入了对 4 岁及以上依赖轮椅的患者的加速许可。[19]
微量肌营养不良蛋白水平与从躺在地板上站起的时间之间的联系的替代指标是 CBER 主任 2024 年决策备忘录中的一个主要因素。
然而,由于斜率较浅(肌肉微肌营养不良蛋白水平每变化一个百分点,就会产生 -0.012 秒),这种联系在视觉上(图形上)并不令人信服,在统计上也不显著(p=0.1388)。
此外,大量数据点与估计的回归线存在较大偏差。
由于第二项关键试验中只有 25% 个体被纳入了此次分析,FDA 专家明确指出,在评估微肌营养不良蛋白剂量反应数据时,应“谨慎解读”结果。因此,尽管使用微肌营养不良蛋白基因插入治疗 DMD 具有科学依据,但临床试验并未表明基因疗法有任何积极作用。[20]
事实上,在辉瑞公司开发的 DMD 基因疗法最近的 3 期试验中,DMD 患者的肌肉中出现了“大量”微肌营养不良蛋白表达,但没有相应的临床改善。据报道,辉瑞公司已停止了对该疗法的研究。[21]
家庭评论
直接受到 DMD 影响的人也表达了类似的担忧。对 delandistrogene moxeparvovec-rokl 安全性和有效性的担忧导致一位家长在 2024 年 9 月写道,他们选择不为孩子进行治疗。[22]
在一段在线视频中,另一位家长批评 Sarepta 研发的治疗方法不够完善,很快就会过时。作为回应,托管该视频的家长支持小组被指控在 Sarepta Therapeutics 威胁停止资助该小组后,从其网站上删除了视频批评。[23]
与临床试验相比,Elevidys 的成本是否值得?
从临床试验结果来看,Elevidys基因治疗300万美元的价格合理吗?[阅读更多: 杜氏肌营养不良症:治疗和费用]
考虑到使用 Elevidys 后体内产生的肌营养不良蛋白的量以及临床试验中检查的北极星门诊评估 (NSAA) 分数,300 万美元对于这种治疗方法来说是非常昂贵的。
我们呼吁Elevidys的开发商Sarepta和全球营销权威公司罗氏: 立即降低为此商业目的设定的高价!
了解更多: 杜氏肌营养不良症的潜在新基因疗法
首次公布死亡消息
2025 年 3 月 18 日,Sarepta Therapeutics 表示,其针对杜氏肌营养不良症的基因疗法 Elevidys 导致的首例死亡病例发生在一名 16 岁男孩身上。[阅读更多]
结论
一位中心主任否决了负责评估该药物的 FDA 科学团队的意见,FDA 批准 delandistrogene moxeparvovec-rokl 作为 DMD 的基因疗法。
Delandistrogene moxeparvovec-rokl 的两项关键试验未能实现其主要临床目标。用于挽救 delandistrogene moxeparvovec-rokl 应用的指标,例如次要终点(例如 10 米步行/跑步测试)和微肌营养不良蛋白水平的间接替代指标,本身就存在问题。
因此,我们认为 FDA 领导层开创了一个危险的先例,未能保护公众免受昂贵且对身体有害的无效药物的侵害。 [24]
了解更多: 杜氏肌营养不良症市场不断增长:但并非所有家庭都能获得治疗
对FDA裁决的一种反应是说问题在于审批标准薄弱,而更严格的审批标准和上市后程序才是解决方案。[25]
FDA 的内部流程应该进行改变,使担任领导职务的个别官员更难以推翻负责监管决策的机构科学家的共识评估,尽管我们同意需要更严格的批准标准和上市后要求。
据在做出两项批准决定时担任 FDA 专员的卡利夫 (Califf) 称,反对该决定的人对于开发治疗严重和罕见疾病的疗法的困难“有着非常简单的临床证据看法”。[26]
我们不同意这种评价。由于 delandistrogene moxeparvovec-rokl 的批准,FDA 应该加强其对新药的决策过程,无论这些新药是用于治疗常见疾病还是罕见疾病。
了解更多: DMD WarrioR 土耳其代表分享对 Elevidys 基因疗法的看法
在离职前的一次采访中,卡利夫还谈到了这一决定,他指出,患有如此严重和罕见疾病的家庭当然渴望任何希望,而且作为政治任命者,他也不愿意推翻职业 FDA 员工做出的决定。虽然这些因素很重要,但不足以支持 FDA 批准 delandistrogene moxeparvovec-rokl 的决定。[27]

{kind=link}