
¿Qué tan efectiva es la terapia génica ELEVIDYS (delandistrógeno moxeparvovec-rokl), que se ha introducido en el mercado farmacéutico, en relación con su costo de 3 millones de dólares estadounidenses? La revisión de los ensayos clínicos de la terapia génica Elevidys será el tema principal de este ensayo. Evaluaremos el costo y la efectividad de esta terapia génica, que fue desarrollada para tratar la distrofia muscular de Duchenne. ¿Vale la pena el costo del Elevidys en comparación con los ensayos clínicos?
Tabla de contenido
Descripción general de los ensayos clínicos
La Administración de Alimentos y Medicamentos (FDA) autorizó una terapia genética revolucionaria en 2023 y 2024 para la distrofia muscular de Duchenne, una enfermedad rara pero mortal de inicio infantil.

Sólo los niños de 4 a 5 años que pudieran caminar (ambulatoriamente) fueron elegibles para el permiso de 2023.
El fallo de 2024 amplió esa autorización para cubrir la aprobación convencional para pacientes ambulatorios con la afección y una mutación verificada en el gen DMD que tengan 4 años de edad o más.
Además, la FDA estableció una aprobación acelerada para personas de 4 años o más que dependen de sillas de ruedas.
Ninguna aprobación estuvo respaldada por resultados de ensayos clínicos apropiados que mostraran que los niños con DMD conservaban o recuperaban la capacidad motora gruesa, ni por evaluaciones científicas positivas del FDA.
Un director de FDA ignoró al personal científico de la agencia, lo que resultó en ambas aprobaciones.
En este artículo se analizan las dos aprobaciones mal pensadas y se indican las razones por las que los líderes del FDA no deberían seguir su ejemplo en el futuro. También se ofrecen sugerencias sobre cómo el FDA puede modificar la forma en que maneja las decisiones regulatorias polémicas.
Cómo el Elevidys fue aprobado por el FDA
Los dos ensayos clínicos aleatorizados de fase 3 del delandistrógeno moxeparvovec-rokl (Elevidys) como cura de reemplazo genético para la distrofia muscular de Duchenne (DMD) se informaron en Nature Medicine el 29 de octubre de 2024. [1] La eficacia clínica de este medicamento no ha sido demostrada de forma concluyente por dos razones, según dicho informe.
Ninguno de los dos ensayos alcanzó sus criterios de valoración principales en cuanto a beneficio clínico. El beneficio clínico se evaluó utilizando una escala estandarizada que mide la trayectoria del desempeño motor grueso, que incluye estar de pie, caminar, saltar y levantar la cabeza.
En segundo lugar, los criterios de valoración alternativos que fueron marginalmente favorables (empleados en última instancia por la Administración de Alimentos y Medicamentos (FDA) para respaldar la aprobación) no pudieron verificarse mediante pruebas estadísticas.
Además, el resultado alternativo primario fueron los niveles de la proteína microdistrofina, un biomarcador que recientemente no mostró relevancia clínica en otro ensayo de terapia génica para DMD. [2]
¿Cuándo se aprobó el Elevidys?
A pesar de la evidencia clínica limitada, el 22 de junio de 2023, delandistrógeno moxeparvovec-rokl recibió la aprobación FDA como la primera terapia de reemplazo genético para la distrofia muscular de Duchenne (DMD), específicamente para niños ambulatorios de 4 a 5 años. [3]
La aprobación se obtuvo a través del programa de aprobación acelerada del FDA. [4]
El 20 de junio de 2024, la FDA amplió su aprobación para incluir la autorización tradicional para personas ambulatorias de 4 años o más diagnosticadas con la enfermedad y que poseen una mutación confirmada en el gen DMD. La agencia también ha otorgado la aprobación acelerada para personas de 4 años o más que dependen de sillas de ruedas.5]
En el documento de revisión clínica del FDA se observó una declaración preocupante relacionada con la acción de aprobación ampliada: [6]
El Dr. Peter Marks, Director del Centro de Evaluación e Investigación Biológica (CBER), aprobó la solicitud anulando la recomendación del equipo de revisión.
El delandistrógeno moxeparvovec-rokl ya está aprobado para su comercialización en Estados Unidos. Recientemente, el precio de la terapia se ha fijado en más de 1 millón de dólares por cura. [7]
La distrofia muscular de Duchenne (DMD) es una enfermedad rara, progresiva y mortal que se caracteriza por la escasez de alternativas de cura efectivas.
Las mutaciones de la distrofia muscular de Duchenne (DMD) son principalmente recesivas y ligadas al cromosoma X, lo que resulta en un impacto predominante en los varones. La incidencia de nacimientos en varones es de aproximadamente 1 en 3600. [Leer más: ¿Qué es DMD?]
Estas mutaciones provocan la degradación del músculo esquelético y cardíaco, que se manifiesta en la primera infancia, a pesar de que el desarrollo musculoesquelético es por lo demás normal. Las personas con un genotipo de DMD suelen perder la capacidad de caminar al principio de la adolescencia y su esperanza de vida suele concluir a los treinta años.
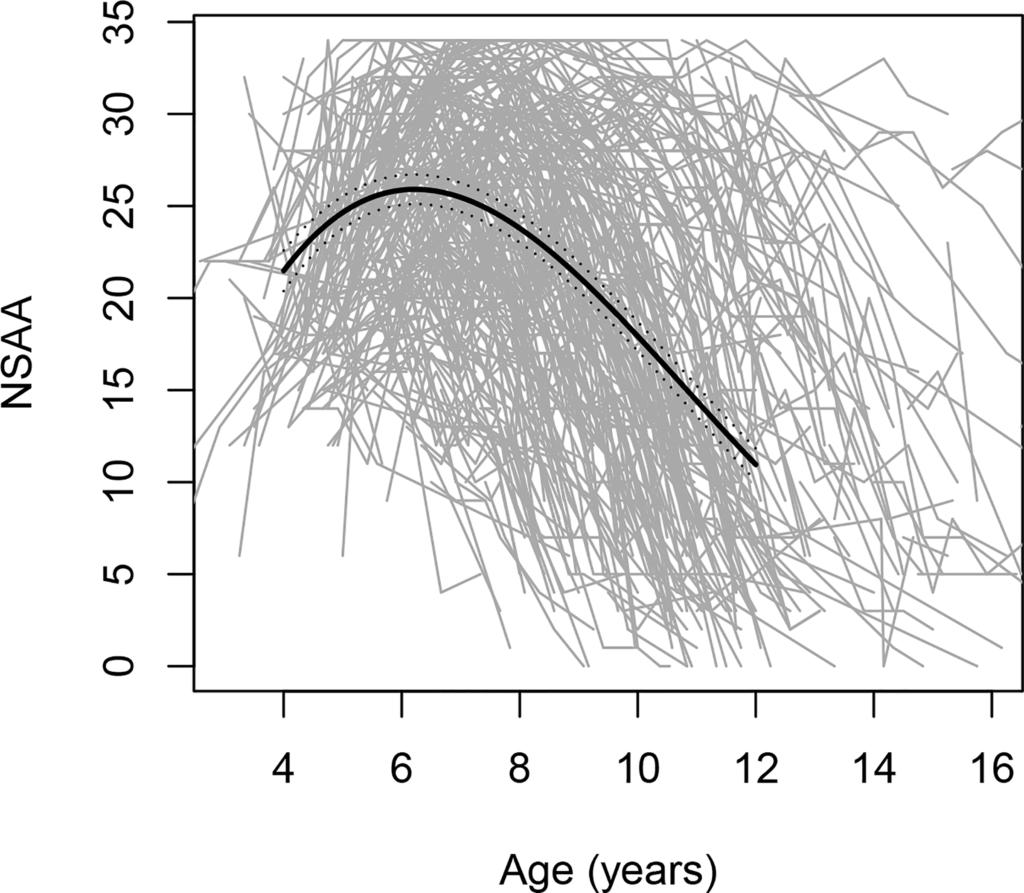
La evolución de la enfermedad es heterogénea, como lo demuestran los datos del FDA que indican una variabilidad significativa en las tendencias de la función motora entre los niños con DMD de 4 a 16 años (ver figura).
Cifra. Cambios en las puntuaciones de la Evaluación Ambulatoria North Star (NSAA), una escala de rendimiento motor grueso, con la edad; incluye 395 trayectorias individuales, cada una representada por una línea gris.
Curas para la DMD de Sarepta
Delandistrógeno moxeparvovec-rokl representa la terapia inaugural modificadora de la enfermedad para la mayoría de los casos de distrofia muscular de Duchenne (DMD); sin embargo, el FDA había aprobado anteriormente cuatro medicamentos para variantes menos comunes de DMD.
Eteplirsen (Exondys 51) es un fármaco bajo escrutinio, diseñado para abordar mutaciones específicas de DMD mediante el uso de oligonucleótidos antisentido que permiten una “saltada de exones” dirigida, evitando así las regiones mutadas en la expresión de gen a proteína. [8]
Hay cinco curas aprobadas por FDA para la DMD, incluido delandistrógeno moxeparvovec-rokl, todas las cuales utilizan formas truncadas de la proteína muscular esencial distrofina (como se detalla a continuación). [9]
Todas estas curas recibieron la aprobación a través del programa de aprobación acelerada del FDA. A pesar de que se han utilizado en pacientes durante varios años, la mayoría de estas curas no han demostrado ventajas clínicas significativas sobre el placebo.
Además, cuatro de los tratamientos, incluido delandistrógeno moxeparvovec-rokl, son producidos por una sola empresa, Sarepta Therapeutics. [10]
El Dr. Robert Califf, comisionado de FDA durante la aprobación de eteplirsen y delandistrógeno moxeparvovec-rokl, expresó sus inquietudes actuales con respecto a los productos DMD de Sarepta en 2024, y afirmó: "Sarepta es como una maldición para mí". [11]
Mecanismo de acción del Elevidys
La expresión de un gen que codifica la proteína distrofina se reduce significativamente o se silencia en mutaciones que causan DMD.
Muchos componentes bioquímicos de la proteína atraviesan las membranas de las células musculares. Además, actúa como amortiguador, manteniendo los músculos fuertes frente al posible deterioro provocado por los frecuentes ciclos de contracción-expansión.12]
En teoría, la DMD se puede reducir o incluso revertir estimulando la expresión de distrofina en las células musculares de quienes la padecen. El delandistrógeno moxeparvovec-rokl, cuyo componente activo es una secuencia de ARN modificada que codifica una variante acortada de la distrofina conocida como “microdistrofina”, se desarrolló en respuesta a esta idea.
La microdistrofina tiene aproximadamente 30% del tamaño de la distrofina no mutada. La proteína acortada observada en pacientes con el tipo más leve de distrofia muscular Becker sirvió como base para la invención de la microdistrofina codificada por el delandistrógeno moxeparvovec-rokl.
Como la secuencia de ARN tenía que estar agrupada en cápsides virales para poder ser administrada al cuerpo, el gen de la distrofina tuvo que ser truncado. El ARN de la distrofina completo supera el límite de capacidad de dichas cápsides.
El delandistrógeno moxeparvovec-rokl se creó mediante una compleja técnica de bioingeniería que da como resultado una proteína que, como mucho, solo restauraría parcialmente la función de la distrofina en todo el cuerpo. La eficacia de esta compleja terapia aún no se ha demostrado en ensayos clínicos.
Aprobación acelerada de 2023
Cuarenta y un niños ambulatorios con DMD, de 4 a 7 años de edad, participaron en el ensayo fundamental para la primera aprobación de delandistrógeno moxeparvovec-rokl. Fueron asignados aleatoriamente 1:1 para recibir el medicamento o un placebo.
El principal resultado clínico predeterminado fue el cambio en el rendimiento motor grueso durante un período de 48 meses, medido mediante una escala estandarizada de 17 ítems; los pacientes tratados con delandistrógeno moxeparvovec-rokl no mejoraron significativamente su rendimiento en comparación con el grupo de control. [13]
Basándose en el criterio de valoración sustituto de la expresión de microdistrofina en el tejido muscular 12 semanas después de la cura con terapia génica, Sarepta, el patrocinador, solicitó una aprobación rápida debido al resultado desfavorable del ensayo clínico.
A pesar de que se observó la expresión de microdistrofina a las 12 semanas, no hubo correlación entre los niveles de microdistrofina y la función muscular resultante. En consecuencia, los revisores científicos de FDA llegaron a la conclusión de que no había ningún beneficio terapéutico en el tratamiento de la DMD a partir del ensayo delandistrógeno moxeparvovec-rokl. Según la revisión clínica de FDA de 2023: [14]
Por lo tanto, incluso para una población pequeña, como los pacientes ambulatorios con DMD de 4 a 5 años que tienen una mutación de DMD confirmada en su gen DMD, estos datos son insuficientes para apoyar la expresión de la microdistrofina de ELEVIDYS como un criterio de valoración sustituto “razonablemente probable para predecir el beneficio clínico” para la aprobación acelerada de ELEVIDYS.
El director del Centro de Evaluación e Investigación Biológica del FDA ignoró a los revisores científicos de la agencia y decidió otorgar una aprobación rápida en niños ambulatorios de 4 y 5 años con DMD a pesar de esta dura crítica.
Los datos de 8 de los 41 pacientes sirvieron como base principal para la decisión. Además, se exigió al patrocinador, como condición para la aprobación acelerada, que finalizara un segundo ensayo aleatorio, cuyos resultados se esperaban para el otoño de 2023, según el fallo. [15]
Aprobación ampliada en 2024
Sarepta dijo en un comunicado de prensa fechado el 30 de octubre de 2023 que no se cumplió el criterio de valoración principal del segundo ensayo aleatorizado fundamental del delandistrógeno moxeparvovec-rokl.
En ese estudio controlado con placebo, 125 pacientes de entre 4 y 7 años fueron asignados aleatoriamente 1:1 para recibir un placebo o el delandistrógeno moxeparvovec-rokl. [16]
El cambio a las 52 semanas en la misma escala de función muscular utilizada en el experimento anterior fue el criterio principal de valoración. Solo se encontró una diferencia promedio de 0,65 puntos entre el medicamento y el placebo en la escala funcional motora gruesa estandarizada (que tiene un rango máximo de 34 puntos), lo que hace que los resultados no sean significativos (p = 0,25).
El FDA debería haber dejado de aprobar el delandistrógeno moxeparvovec-rokl en ese momento. En cambio, para la aprobación de 2023, el fabricante hizo hincapié en los resultados secundarios que, según ellos, produjeron “resultados sólidos y estadísticamente significativos… [que] respaldan la eficacia de un suplemento”.
Los revisores científicos de FDA se mostraron muy críticos con estos nuevos hallazgos. En particular, las pruebas de caminata/carrera de 10 metros y el tiempo que se tardaba en levantarse del suelo eran dos resultados secundarios importantes.
Los sujetos que tomaron Delandistrógeno moxeparvovec-rokl realizaron estas actividades en promedio 0,5 segundos más rápido que los sujetos placebo, quienes tardaron 3,5 y 4,9 segundos al inicio, respectivamente.
Estos hallazgos, que no fueron preespecificados ni corregidos estadísticamente para los múltiples análisis de datos, “no pueden respaldar la eficacia de ELEVIDYS”, según los revisores de FDA.
Los datos, según los revisores de FDA, también fueron “engañosos y no pueden orientar a ninguna de las partes interesadas, incluidos pacientes, familiares y cuidadores, y prescriptores, a tomar decisiones informadas sobre los posibles beneficios de la cura con ELEVIDYS”. [17]
Sin embargo, en junio de 2024, el director del CBER anuló la decisión de su equipo y aprobó el delandistrógeno moxeparvovec-rokl por segunda vez, a pesar de las críticas desfavorables de los científicos del FDA. [18]
Como se mencionó anteriormente, la segunda aprobación amplió y fortaleció la autorización para incluir la aprobación típica en pacientes ambulatorios con la afección que tengan 4 años de edad o más y tengan una mutación verificada en el gen DMD. Además, en la segunda aprobación se incorporó la autorización acelerada para personas dependientes de sillas de ruedas de 4 años de edad o más. [19]
La indicación sustitutiva de la conexión entre los niveles de proteína microdistrofina y el tiempo necesario para levantarse después de estar acostado en el suelo fue un factor importante en el memorando de decisión de 2024 del Director del CBER.
Sin embargo, este vínculo no fue convincente visualmente (gráficamente) ni estadísticamente significativo (p = 0,1388) debido a la pendiente suave (-0,012 segundos por cada cambio porcentual en el nivel de microdistrofina del músculo).
Además, un gran número de puntos de datos se desviaron significativamente de la línea de regresión estimada.
Dado que en el segundo ensayo fundamental sólo se incluyeron 25% de los individuos en este análisis, los expertos de FDA habían señalado explícitamente que los resultados debían “interpretarse con cautela” al evaluar los datos de respuesta a la dosis de microdistrofina. Como resultado, a pesar de la justificación científica para utilizar la inserción del gen de microdistrofina para tratar la DMD, los ensayos clínicos no han demostrado que la terapia génica tenga efectos positivos. [20]
De hecho, en un reciente ensayo de fase 3 de una terapia génica para la DMD desarrollada por Pfizer se observaron “cantidades significativas” de expresión de microdistrofina en los músculos de pacientes con DMD, pero no hubo una mejoría clínica proporcional. Según los informes, Pfizer ha dejado de trabajar en su cura. [21]
Comentarios de la familia
Preocupaciones similares han sido expresadas por aquellos que se ven directamente afectados por la DMD. Las inquietudes con respecto a la seguridad y eficacia del delandistrógeno moxeparvovec-rokl llevaron a un padre a escribir en septiembre de 2024 que había decidido no seguir la terapia para su hijo. [22]
En un vídeo en línea, otro padre criticó a Sarepta por crear una cura que era insuficiente y que pronto se volvería obsoleta. En respuesta, un grupo de apoyo para padres que alojaba el vídeo fue acusado de eliminar la crítica en vídeo de delandistrógeno moxeparvovec-rokl de su sitio web después de que Sarepta Therapeutics amenazara con dejar de financiar al grupo. [23]
¿Vale la pena el costo del Elevidys en comparación con los ensayos clínicos?
Teniendo en cuenta los resultados de los ensayos clínicos, ¿es razonable el precio de 3 millones de dólares por la terapia génica Elevidys? [Leer más: Distrofia muscular de Duchenne: tratamiento y costo]
Teniendo en cuenta la cantidad de distrofina producida en el cuerpo después del uso de Elevidys y los puntajes de la Evaluación Ambulatoria North Star (NSAA) examinados en ensayos clínicos, 3 millones de dólares es muy exorbitante para este método de tratamiento.
Nuestro llamamiento a Sarepta, el desarrollador de Elevidys, y a Roche, la autoridad mundial en marketing: ¡Reduzca inmediatamente este alto precio establecido para este propósito comercial!
Más información: Posibles nuevas terapias genéticas para la distrofia muscular de Duchenne
Primer anuncio de muerte
El 18 de marzo de 2025, Sarepta Therapeutics dijo que la primera muerte registrada asociada con su terapia genética para la distrofia muscular de Duchenne, Elevidys, había ocurrido en un niño de 16 años.Leer más]
Conclusión
Un director del centro anuló la decisión del equipo científico del FDA, que estaba encargado de evaluar el medicamento, y el FDA aprobó el delandistrógeno moxeparvovec-rokl como terapia genética para la DMD.
Los dos ensayos fundamentales de delandistrógeno moxeparvovec-rokl no lograron cumplir con sus principales objetivos clínicos. Los indicadores utilizados para rescatar la aplicación de delandistrógeno moxeparvovec-rokl, como los criterios de valoración secundarios (por ejemplo, pruebas de caminata/carrera de 10 metros) y el indicador indirecto sustituto de los niveles de microdistrofina, fueron problemáticos en sí mismos.
Como resultado, creemos que el liderazgo de FDA ha creado un precedente riesgoso que no protege al público de estar expuesto a un medicamento ineficaz que es costoso y físicamente exigente. [24]
Más información: El mercado de la distrofia muscular de Duchenne crece: pero no todas las familias tienen acceso a los tratamientos
Una forma de reaccionar ante la decisión del FDA es decir que el problema son los criterios de aprobación débiles y que la solución son normas de aprobación y procedimientos posteriores a la comercialización más estrictos.25]
Los procesos internos del FDA deberían modificarse para que sea mucho más difícil para los funcionarios individuales en roles de liderazgo anular la evaluación de consenso de los científicos de la agencia asignados a una decisión regulatoria, aun cuando estamos de acuerdo en que se requieren estándares de aprobación y requisitos posteriores a la comercialización más estrictos.
Según Califf, el Comisionado FDA en el momento de las dos decisiones de aprobación de delandistrógeno moxeparvovec-rokl, quienes se oponen a la decisión tienen una “visión muy simplista de la evidencia clínica” sobre las dificultades para desarrollar terapias para trastornos graves y poco comunes. [26]
No estamos de acuerdo con esa evaluación. El FDA debería mejorar su proceso de toma de decisiones sobre nuevos fármacos, ya sean para enfermedades comunes o poco comunes, como resultado de la aprobación del delandistrógeno moxeparvovec-rokl.
Más información: El representante turco de DMD WarrioR comparte su opinión sobre la terapia génica Elevidys
En una entrevista previa a su partida, Califf también habló sobre la decisión, señalando que las familias con enfermedades tan graves y poco comunes estaban comprensiblemente ansiando cualquier esperanza, y que como designado político, también se mostraba reacio a revocar las decisiones tomadas por los empleados de carrera de FDA. Si bien estos factores son significativos, no son suficientes para respaldar la decisión de FDA de aprobar el delandistrógeno moxeparvovec-rokl. [27]

{kind=link}