









 FDA Ablehnungsschreiben im Jahr 2019")
Die US-amerikanische Lebensmittel- und Arzneimittelzulassungsbehörde FDA veröffentlichte am Donnerstag über 200 Briefe, die sie an Unternehmen geschickt hatte, nachdem deren Medikamente abgelehnt worden waren. Damit lenkte sie die Aufmerksamkeit auf einen Teil des Arzneimittelprüfungsprozesses, der manchmal übersehen wird. Einer dieser Ablehnungsbriefe betraf Vyondys 53 (Golodirsen), ein von Sarepta Therapeutics hergestelltes und für die Exon-53-Skipping-Therapie entwickeltes Medikament.
Die Behörde konzentrierte sich ausschließlich auf Briefe an Hersteller, deren Produkte schließlich zugelassen wurden. Die meisten davon waren bereits im Laufe der Jahre auf den Markt gekommen. Durch die Zusammenstellung aller Briefe an einem Ort behauptete die Behörde, dies sei Teil einer umfassenderen Anstrengung zur Förderung der Transparenz.

Inhaltsverzeichnis
Details zum Ablehnungsschreiben FDA zu Vyondys 53
Das FDA erteilte 2019 seinen Ablehnungsbescheid für Vyondys 53 (Golodirsen), eine Exon-53-Skipping-Behandlung für die Duchenne-Muskeldystrophie. Im FDA hieß es, dass der klinische Nutzen von Vyondys 53 angesichts des „geringen Anstiegs des verkürzten Dystrophinspiegels“, der durch die Behandlung zustande komme, wahrscheinlich „entsprechend gering“ sei.
Dystrophinspiegel um 9 Tausendstel erhöht
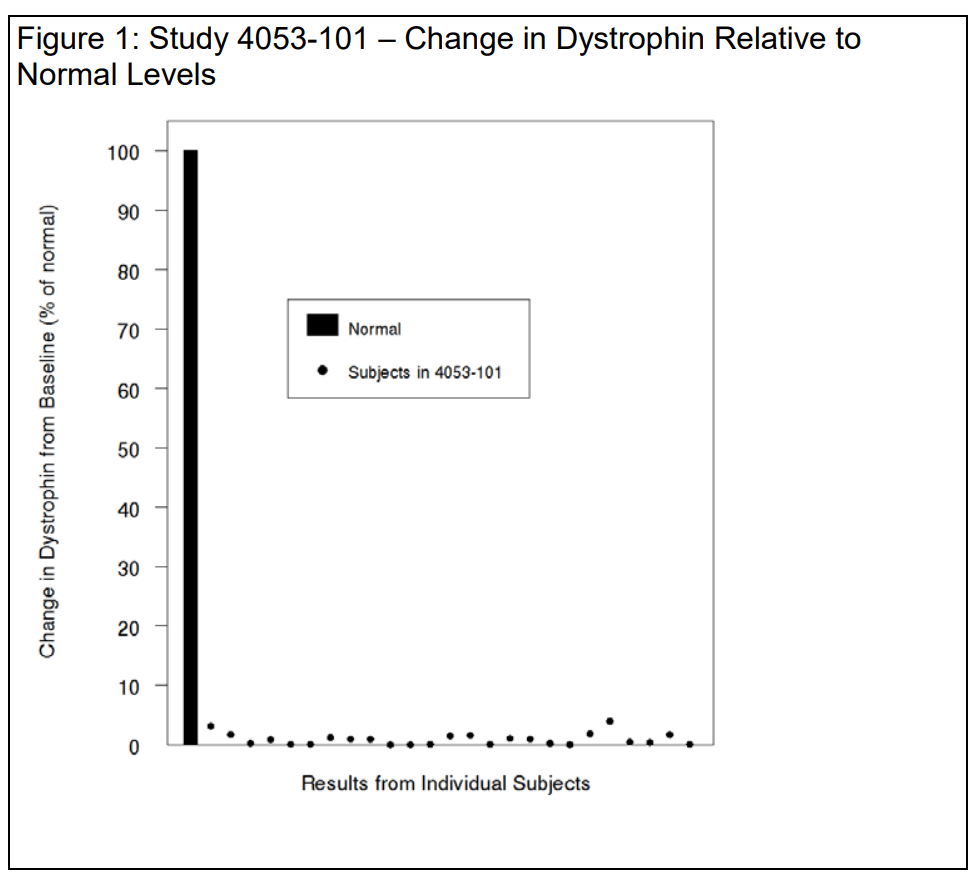
Im Ablehnungsschreiben des FDA für Vyondys 53 (Golodirsen) wurde ein sehr geringer Anstieg des Dystrophinspiegels nach der Einnahme des Medikaments festgestellt.:
Bei den 25 auswertbaren Patienten der Studie lag der mittels Western Blot ermittelte mittlere Dystrophin-Ausgangswert bei 0,10 ± 0,07 (Prozent des Normalwerts; Mittelwert ± SD). In Woche 48 lag der mittlere Dystrophin-Wert bei 1,02 ± 1,03 (Prozent des Normalwerts; Mittelwert ± SD), was einem mittleren Anstieg von 0,92 ± 1,01 Prozent des Normalwerts entspricht.
Die individuellen Veränderungen des Dystrophins sind unten in Abbildung 1 dargestellt. Dabei wird eine Normalskala (100%) verwendet, um die Daten in die richtige Perspektive zu rücken und eine Übertreibung der Effektstärke zu vermeiden. Sie stellen fest, dass die mittleren Anstiege des trunkierten Dystrophins bei Patienten mit Mutationen, die für Exon-53- bzw. Exon-51-Skipping anfällig sind, in Reaktion auf Golodirsen und Eteplirsen ähnlich sind – mit absoluten mittleren Anstiegen von 0,9%, also 9 Tausendstel. Dem stimmen wir zu.
Wenn man davon ausgeht, dass ein geringer Anstieg des verkürzten Dystrophins in der Größenordnung von 9 Promille mit hinreichender Wahrscheinlichkeit einen klinischen Nutzen vorhersagt, liegt die Annahme nahe, dass der klinische Nutzen entsprechend gering ausfällt.
Klinische Wirkung von Vyondys 53
Die Daten zum 6-Minuten-Gehtest aus Studie 4053-101 werden hier nicht diskutiert, zeigen aber einen fortschreitenden Verlust körperlicher Funktionen bei nahezu allen Jungen. Zudem besteht kein Zusammenhang zwischen der Aufrechterhaltung der körperlichen Leistungsfähigkeit und dem Ausmaß der reduzierten Dystrophinproduktion. Dies deutet darauf hin, dass, falls Golodirsen tatsächlich einen klinischen Effekt hat, dieser nur gering ist.
Schwerwiegende Nebenwirkungen von Vyondys 53, laut FDA
Im Dezember 2019 genehmigte die FDA Sareptas Vyondys 53 als erste zielgerichtete Therapieoption für DMD-Patienten, die anfällig für Exon-53-Skipping sind. Das Medikament war jedoch vier Monate zuvor von der FDA in einem CRL abgelehnt worden, das von Ellis Unger, dem damaligen Leiter des Office of Drug Evaluation des Center for Medicine Evaluation and Research, unterzeichnet worden war.
In der Begründung der FDA hieß es, angesichts des „geringen Anstiegs des verkürzten Dystrophins“, den die Behandlung hervorrief, sei der therapeutische Effekt von Vyondys 53 wahrscheinlich „entsprechend gering“. Unger erklärte weiter, es gebe „keinen Zusammenhang“ zwischen der durch Vyondys 53 bei den Jungen produzierten Dystrophinmenge und ihrer körperlichen Leistungsfähigkeit. Außerdem hätten „praktisch alle“ Jungen, die nach der Verabreichung des Medikaments untersucht wurden, einen zunehmenden Verlust körperlicher Funktionen gezeigt.
Als ob die blauen Flecken nicht genug wären, zählte Unger eine lange Liste von Sicherheitsbedenken auf, mit denen die 53 Vyondys-Nutzer zu kämpfen hatten. Dazu gehörten „Nierentoxizität“ und „schwere Infektionen im Zusammenhang mit der Medikamentenverabreichung“. Er behauptet, beides könne „tödlich sein“, wobei Letzteres „schwer oder unmöglich zu überwachen“ sei.
Angesichts der beiden jüngsten Todesfälle durch schweres Leberversagen im Zusammenhang mit Sareptas Gentherapie Elevidys ist diese zweite Warnung besonders besorgniserregend. Wie Unger in dem Brief anmerkte, wurden beide Wege mit Leberschäden in Verbindung gebracht, obwohl es sich bei Vyondys 53 um ein Antisense-Oligonukleotid (ASO) und nicht um eine Gentherapie auf Basis von Adeno-assoziierten Viren (AAV) wie Elevidys handelt.
Mehr lesen: Vyondys 53 FDA Ablehnungsschreiben
Wie wurde Vyondys 53 genehmigt?
Um die Bedenken der Aufsichtsbehörde im CRL auszuräumen, legte Sarepta später Berufung ein und traf sich mit dem FDA. Dies führte schließlich dazu, dass der damalige Leiter des Office of New Drugs des FDA, Peter Stein, der die Behörde im April verließ, Vyondys 53 genehmigte.
Pharmaunternehmen veröffentlichen nicht alle Informationen
„Viel zu lange haben Arzneimittelentwickler bei der Entwicklung des FDA-Programms nur aufs Spiel gesetzt“, sagte Dr. Marty Makary, MPH, Kommissar für FDA. „Arzneimittelentwickler und Kapitalmärkte wünschen sich gleichermaßen Vorhersehbarkeit. Heute sind wir der Bereitstellung dieser Informationen einen Schritt näher gekommen, mit dem ultimativen Ziel, Patienten schneller Heilmittel und wirksame Behandlungen zur Verfügung zu stellen.“
Da die FDA-Zulassungsbehörde in der Vergangenheit keine CRLs für anhängige Anträge veröffentlicht hat, stellen Sponsoren die Gründe für ihre Entscheidung gegenüber ihren Stakeholdern und der Öffentlichkeit häufig falsch dar. Laut einer 2015 von Forschern der FDA-Zulassungsbehörde durchgeführten Analyse vermieden es Sponsoren, 85 der Bedenken der FDA-Zulassungsbehörde hinsichtlich Sicherheit und Wirksamkeit zu erwähnen, als sie die Ablehnung ihres Antrags öffentlich bekannt gaben. Darüber hinaus werden diese wichtigen Informationen in etwa 40 der Fälle nicht veröffentlicht, wenn die FDA-Zulassungsbehörde eine neue klinische Studie zur Sicherheit oder Wirksamkeit fordert. Auch die aus Ablehnungen gezogenen Lehren werden branchenintern nicht geteilt, was dazu führt, dass Unternehmen immer wieder ähnliche Fehler machen. – Weiterlesen: FDA setzt auf radikale Transparenz durch die Veröffentlichung vollständiger Antwortschreiben –

{kind=link}