
İlaç geliştirme süreci, ilk keşiften pazara sunulmasına kadar genellikle 10 ila 15 yıl arasında süren oldukça karmaşık, uzun ve maliyetli bir yolculuktur. Duchenne Musküler Distrofisi için tedaviler de dahil olmak üzere, her biri yeni bir ilacın genel halka sunulmadan önce güvenliğini, etkinliğini ve potansiyel risklerini değerlendirmek için dikkatlice tasarlanmış birkaç aşamayı içerir. İlaç geliştirmenin aşamaları, Gıda ve İlaç Dairesi (FDA) ve Avrupa İlaç Ajansı (EMA) gibi düzenleyici kurumların birincil odak noktası olan klinik öncesi araştırma ve klinik denemelere ayrılır.
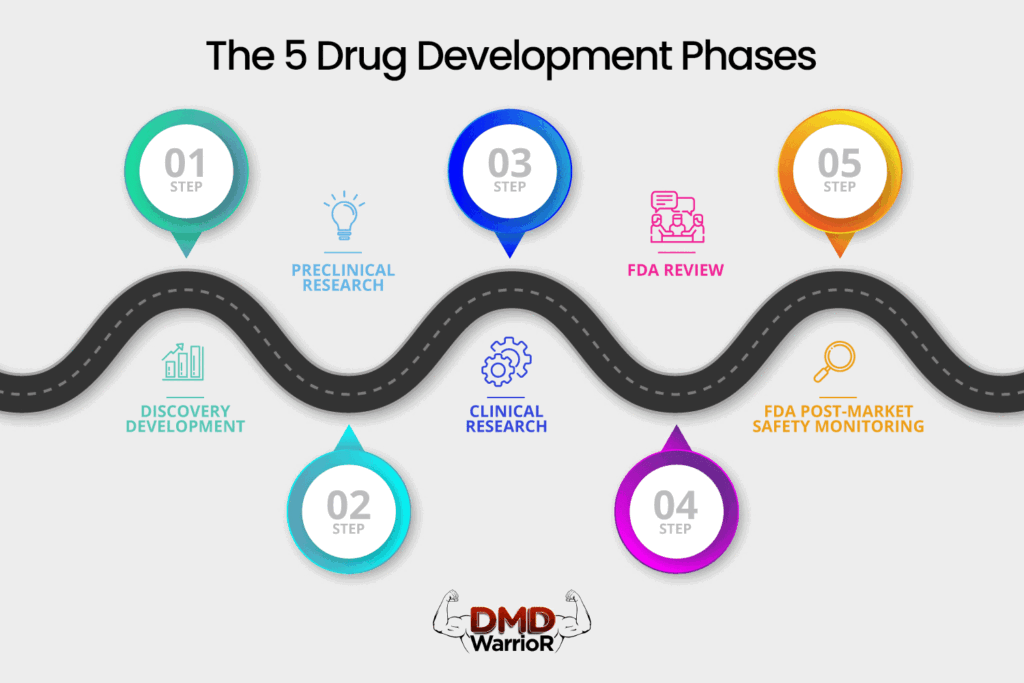
Yeni bir ilacın "başarılı" sayılabilmesi için beş belirgin aşamadan geçmesi gerekir: 1) Keşif ve Geliştirme; 2) Klinik Öncesi Araştırma; 3) Klinik Denemeler; 4) FDA İlaç İncelemesi; ve 5) FDA Piyasa Sonrası Güvenlik İzleme.FDA]

İçindekiler
Keşif ve Geliştirme
Bir ilaç klinik deneme aşamasına girmeden önce, önemli miktarda ön araştırma gerçekleşir. Bu aşama şunları içerir:
- İlaç Keşfi: İlk adım, bir hastalıkta rol oynayan biyolojik bir hedefi (örneğin bir protein veya gen) tanımlamayı içerir. Bilim insanları, hedefi etkileyebilecek adayları belirlemek için büyük bileşik kütüphanelerini tarayabilir. Bu bileşikler daha sonra etkinliklerini artırmak ve olası yan etkileri azaltmak için optimize edilir.
Klinik Öncesi Araştırma
- Klinik Öncesi: Umut vadeden bir bileşik tanımlandıktan sonra, toksisitesini, farmakokinetiğini (ilacın vücutta nasıl emildiği, dağıtıldığı, metabolize edildiği ve atıldığı) ve farmakodinamiğini (ilacın vücudu nasıl etkilediği) değerlendirmek için in vitro (canlı bir organizmanın dışında) ve in vivo (canlı organizmalarda, genellikle hayvanlarda) laboratuvar testlerine tabi tutulur. Bu çalışmalar, ilacın insanlarda güvenli ve etkili olma olasılığının belirlenmesine yardımcı olur.
- Toksikoloji Çalışmaları: Klinik öncesi araştırmalar sırasında bilim insanları ilacın olası zararlı etkilerini değerlendirmek için detaylı çalışmalar yürütürler. Amaç, maksimum tolere edilen dozu ve oluşabilecek herhangi bir yan etkiyi belirlemektir.
Klinik öncesi çalışmalar başarılı olursa ilaç, insanlarda denenmeyi içeren klinik araştırma aşamasına geçiyor.
Klinik Denemeler
Faz 1, faz 2, faz 3 klinik çalışmalar nelerdir?
Klinik araştırma süreci, bir ilacın insanlarda güvenliğini, etkinliğini ve genel fayda-risk oranını kademeli olarak değerlendirmek için dört ayrı faza (Faz 1, Faz 2, Faz 3 ve Faz 4) ayrılır. Her faz, değerlendirme sürecinde benzersiz bir amaca hizmet eder.
Aşama 1: Güvenlik ve Dozaj
Faz 1, ilacın klinik öncesi araştırmalardan sonra insanlarda ilk kez test edildiği zamandır. Faz 1 denemelerinin temel amacı, ilacın güvenlik profilini değerlendirmek, güvenli bir dozaj aralığı belirlemek ve herhangi bir yan etkiyi tespit etmektir.
- Çalışma Tasarımı: Tipik olarak, Faz 1 denemeleri küçüktür ve 20 ila 100 sağlıklı gönüllü veya hastalığı/durumu olan kişileri içerir. Bu denemeler genellikle hastane veya araştırma tesisi gibi kontrollü bir klinik ortamda yürütülür.
- Amaçlar:
- Emniyet:Araştırmacılar, hafif semptomlardan daha ciddi reaksiyonlara kadar değişen herhangi bir yan etki açısından gönüllüleri yakından takip ediyor.
- Dozaj:İlacın ilk dozu genellikle muhafazakardır. İlaç, maksimum tolere edilebilir dozu belirlemek için doz artırma olarak bilinen bir işlemde kademeli olarak artırılır.
- Farmakokinetik ve Farmakodinamik:Araştırmacılar ayrıca ilacın vücut tarafından nasıl emildiğini, dağıtıldığını, metabolize edildiğini ve atıldığını ve biyolojik etkilerini de inceliyorlar.
- Süre: Faz 1 denemeleri genellikle birkaç ay sürer. Başarılı olursa, ilaç Faz 2'ye geçer.
Aşama 2: Etkinlik ve Yan Etkiler
Faz 2 çalışmaları, ilacın tedavi etmeyi amaçladığı hastalık veya duruma sahip belirli bir hasta popülasyonunda ilacın etkinliğini ve güvenliğini daha fazla araştırmak için yürütülür.
- Çalışma Tasarımı: Bu denemeler genellikle 100 ila 300 katılımcıyı içeren Faz 1'den daha büyüktür. Odak noktası, ilacın hastalardaki etkinliğini test etmek ve uzun süreli kullanımla ilişkili herhangi bir olumsuz etkiyi belirlemektir.
- Amaçlar:
- Etkinlik: Araştırmacılar, ilacın hedeflenen durumdaki hastalarda istenen terapötik etkiyi üretip üretmediğini belirlemeyi amaçlar. Etkinlik, semptomlarda veya hastalık biyobelirteçlerinde iyileşme gibi klinik sonuçlarla ölçülür.
- Emniyet:1. Faz'a benzer şekilde araştırmacılar güvenliği izlemeye devam ediyor, ancak birincil hedef hastalığı olan hastalarda ortaya çıkabilecek yan etkileri tespit etmek.
- Optimum Doz:2. Fazda araştırmacılar, 1. Fazda elde edilen bilgiler ışığında ilacın doz rejimini daha da geliştirirler.
- Süre: Faz 2 denemeleri genellikle birkaç aydan birkaç yıla kadar sürer. İlaç etkililik ve olumlu bir güvenlik profili gösterirse, Faz 3'e ilerler.
Aşama 3: Doğrulayıcı Denemeler (Etkinlik ve Güvenlik)
Faz 3 denemeleri, ilacın etkinliğini doğrulamak, yan etkilerini izlemek ve mevcut tedavilerle veya plasebo ile karşılaştırmak için tasarlanmış büyük ölçekli çalışmalardır. Bu faz, ilacın düzenleyici otoriteler tarafından kullanım için onaylanıp onaylanmayacağını belirlemede kritik bir rol oynar.
- Çalışma Tasarımı: Faz 3 denemeleri çok daha fazla katılımcıyı içerir, tipik olarak 1.000 ila 3.000 veya daha fazla katılımcıyı içerir. Bu denemeler genellikle çok merkezlidir, yani birden fazla lokasyonda ve hatta bazen uluslararası olarak yürütülürler.
- Amaçlar:
- Etkinlik: Amaç, ilacın mevcut tedavilere veya plaseboya kıyasla klinik olarak anlamlı bir fayda sağladığını doğrulamaktır.
- Emniyet: Araştırmacılar yan etkileri izlemeye devam ediyor, ancak daha büyük örneklem boyutu, daha önceki aşamalarda gözden kaçmış olabilecek daha az yaygın yan etkilerin belirlenmesine yardımcı oluyor.
- Karşılaştırmalı Etkinlik: Faz 3 denemeleri genellikle yeni ilacı üstünlüğünü veya daha aşağı olmadığını göstermek için standart tedavilerle veya plasebo ile karşılaştırır.
- Süre: Faz 3 denemeleri, kapsamlı veri toplama gerektirdiğinden birkaç yıl sürebilir. Bu denemeler genellikle ilaç geliştirme sürecinin en pahalı ve zaman alıcı kısmıdır.
- Sonuç: Eğer bir Faz 3 denemesi başarılı olursa, ilaç sponsoru FDA veya EMA gibi düzenleyici otoritelere Yeni İlaç Başvurusu (NDA) veya Biyolojik Lisans Başvurusu (BLA) sunabilir. Onaylanırsa, ilaç son faza geçer: Aşama 4.
FDA İlaç İncelemesi
Bir ilaç bileşiğinin klinik deneylerden elde edilen güvenlik ve etkinlik sonuçları, faz 1, faz 2, faz 3 ve faz 4 klinik deneylerini geçtikten sonra hekimler, kimyagerler, istatistikçiler, mikrobiyologlar ve farmakologlardan oluşan bir uzman panel tarafından incelenir.
Bir biyoteknoloji veya ilaç şirketi, FDA değerlendirmesi talep etmek için biyolojikler için Biyolojik Lisans Başvurusu (BLA) veya ilaçlar için Yeni İlaç Başvurusu (NDA) dosyalamalıdır. Bundan sonra, FDA başvuruyu onaylamalı ve değerini değerlendirmek üzere bir grup profesyonel belirlemelidir.
Grup, ilacın risk-fayda analizini, olası yan etkileri ve hasta sonuçlarını göz önünde bulundurarak klinik çalışmayı birlikte inceler. FDA, amaçlanan kullanımı için güvenli ve etkili olduğuna karar verilirse ilacın ABD'de üretilmesini, pazarlanmasını ve dağıtımını yetkilendirir.
FDA Pazar Sonrası Güvenlik İzleme
Bir ilaç düzenleyici kurumlar tarafından onaylandıktan sonra, Faz 4 olarak da bilinen pazarlama sonrası faza girer. Bu fazda, ilaç genel nüfus tarafından kullanılabilir hale gelir, ancak araştırmacılar uzun vadeli güvenliğini, etkinliğini ve nadir veya uzun vadeli yan etkilerini izlemeye devam eder.
- Pazarlama Sonrası Gözetim:4. Aşamada, devam eden çalışmalar ve hasta raporları (istenmeyen olay bildirim sistemlerinden gelen raporlar gibi), daha önceki denemelerde tespit edilmemiş olabilecek nadir yan etkilerin belirlenmesine yardımcı olur.
- Gerçek Dünya Verileri: Araştırmacılar ayrıca ilacın gerçek dünya koşullarında ve klinik çalışmalarda yeterince temsil edilmemiş olabilecek çeşitli popülasyonlarda nasıl performans gösterdiğine ilişkin veri topluyorlar.
Daha fazla bilgi edin: Duchenne Tedavileri (Tüm Araştırmaların Listesi)
Sonuç: İlaç Geliştirmede Her Aşamanın Önemi
Klinik ilaç geliştirmenin her aşaması, yeni bir ilacın pazara ulaşmadan önce hem güvenli hem de etkili olduğundan emin olmakta önemli bir rol oynar. Güvenliğin birincil endişe olduğu Aşama 1'den, ilacın genel fayda-risk oranını büyük bir hasta popülasyonunda titizlikle test eden Aşama 3'e ve pazara sunulduktan sonra uzun vadeli etkileri izleyen Aşama 4'e kadar her aşama, ilacın hastaları nasıl etkileyeceği anlayışına katkıda bulunur.
İlaç geliştirme sürecinin tamamı karmaşık bir denge eylemidir, çünkü ilaç şirketleri, düzenleyici kurumlar ve araştırmacılar potansiyel zararı en aza indirirken yenilikçi ve hayat değiştiren tedavileri pazara sunmak için birlikte çalışırlar. İlaç geliştirmeye yatırılan zaman, çaba ve maliyetler önemlidir, ancak bunlar tıp bilimini ilerletmek ve dünya çapında hasta sonuçlarını iyileştirmek için elzemdir.

{kind=link}