









 FDA Rejection Letter in 2019")
Управление по контролю за продуктами питания и лекарственными средствами опубликовало более 200 писем, отправленных компаниям после того, как в четверг их лекарства были отклонены, привлекая внимание к порой упускаемой из виду части процесса рассмотрения лекарств. Одно из таких писем об отказе было отправлено на препарат Vyondys 53 (голодирсен), производимый компанией Sarepta Therapeutics и разработанный для терапии пропуска экзона 53.
Агентство сосредоточилось только на письмах производителям, чья продукция в конечном итоге была одобрена, большинство из которых уже были выпущены в течение многих лет. Собрав все письма в одном месте, агентство заявило, что эта деятельность является частью более широких усилий по повышению прозрачности.

Оглавление
Подробности письма об отказе FDA о Vyondys 53
В 2019 году FDA выдал письмо об отклонении заявки на препарат Vyondys 53 (голодирсен), препарат для лечения мышечной дистрофии Дюшенна, пропускающий экзон 53. В документе FDA говорится, что, учитывая «небольшое повышение уровня укороченного дистрофина», достигаемое в результате лечения, клиническая польза от препарата Vyondys 53, вероятно, будет «соразмерно небольшой».
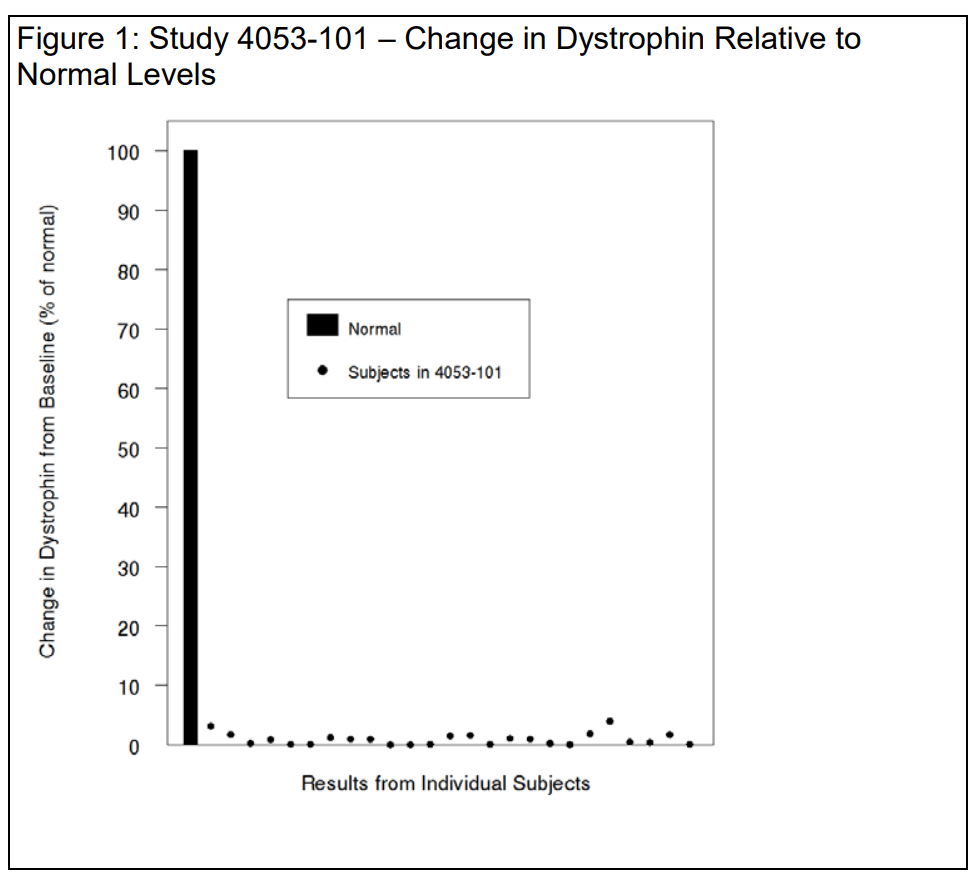
Уровень дистрофина увеличился на 9 частей на тысячу
В письме об отказе FDA от препарата Vyondys 53 (голодирсен) отмечено очень небольшое повышение уровня дистрофина после применения препарата.:
У 25 пациентов, участвовавших в исследовании, исходный средний уровень дистрофина, оцененный методом вестерн-блоттинга, составил 0,10 ± 0,07 (процент от нормы; среднее значение ± СД). На 48-й неделе средний уровень дистрофина составил 1,02 ± 1,03 (процент от нормы; среднее значение ± СД), что соответствует среднему повышению на 0,92 ± 1,01% от нормы.
Индивидуальные изменения уровня дистрофина показаны на рисунке 1 ниже в нормальном масштабе (100%), чтобы представить данные в правильном свете и избежать преувеличения величины эффекта. Вы отмечаете, что среднее увеличение уровня укороченного дистрофина в ответ на голодирсен и этеплирсен схоже у пациентов с мутациями, поддающимися пропуску экзона 53 и экзона 51, соответственно – с абсолютным средним увеличением 0,9%, то есть 9 частей на тысячу. Мы согласны.
Если принять предположение, что небольшое увеличение усеченного дистрофина, порядка 9 частей на тысячу, с достаточной вероятностью предскажет клиническую пользу, то разумно предположить, что клиническая польза будет соразмерно небольшой.
Клинический эффект Виондиса 53
Данные 6-минутной ходьбы из исследования 4053-101 в данной работе не обсуждаются, но они демонстрируют прогрессирующую потерю физической функции практически у всех мальчиков. Более того, не наблюдается корреляции между сохранением физической работоспособности и уровнем продукции укороченного дистрофина, что дополнительно свидетельствует о том, что если голодирсен действительно имеет клинический эффект, то его величина невелика.
Серьёзные побочные эффекты препарата Vyondys 53, согласно FDA
В декабре 2019 года комиссия FDA одобрила препарат Vyondys 53 компании Sarepta в качестве первого варианта таргетной терапии для пациентов с МДД, подверженных пропуску экзона 53. Однако четырьмя месяцами ранее комиссия FDA отклонила препарат в рецензируемом списке, подписанном Эллисом Унгером, который в то время возглавлял Отдел оценки лекарственных средств Центра оценки и исследований лекарственных средств.
В обосновании отказа FDA заявил, что, учитывая «небольшое увеличение укороченного дистрофина», вызванное лечением, терапевтический эффект от препарата Vyondys 53, вероятно, будет «соразмерно малым». Унгер продолжил, объяснив, что «не существует никакой корреляции» между количеством дистрофина, вырабатываемого Vyondys 53 у мальчиков, и их физическими показателями, и что «практически у всех» мальчиков, обследованных после приема препарата, наблюдалась нарастающая потеря физических функций.
Как будто синяков было недостаточно, Унгер перечислил длинный список проблем безопасности, с которыми пришлось столкнуться пользователям Vyondys 53. Среди них были «почечная токсичность» и «серьёзные инфекции, связанные с введением препарата». Он утверждает, что оба эти фактора «могут привести к летальному исходу», причём последние «трудно или невозможно контролировать».
Учитывая два недавних случая смерти от тяжёлой печёночной недостаточности, связанных с генной терапией Elevidys компании Sarepta, это второе предупреждение вызывает особую тревогу. Как отметил Унгер в письме, оба пути введения препарата связаны с поражением печени, несмотря на то, что Vyondys 53 представляет собой антисмысловой олигонуклеотид (ASO), в отличие от генной терапии на основе аденоассоциированного вируса (AAV), такой как Elevidys.
Читать далее: Письмо-отказ от Vyondys 53 FDA
Как был одобрен Vyondys 53?
Чтобы решить вопросы, поднятые регулятором в CRL, компания Sarepta впоследствии подала апелляцию и встретилась с FDA. В конечном итоге это привело к тому, что тогдашний руководитель Управления новых лекарственных средств FDA Питер Штайн, покинувший агентство в апреле, одобрил препарат Vyondys 53.
Фармацевтические компании не публикуют всю информацию
«Слишком долго разработчики лекарств играли в угадайку, ориентируясь в рамках FDA», — заявил комиссар FDA Марти Макари, доктор медицины и магистр общественного здравоохранения. «Разработчики лекарств и рынки капитала одинаково хотят предсказуемости. Поэтому сегодня мы на шаг ближе к тому, чтобы предоставить им эту предсказуемость, и наша главная цель — быстрее предоставлять пациентам лекарства и эффективные методы лечения».
Поскольку FDA исторически воздерживался от публикации списков отозванных лекарственных препаратов (CRL) для находящихся на рассмотрении заявок, спонсоры часто искажают обоснование решения FDA перед своими заинтересованными сторонами и общественностью. Согласно анализу, проведенному исследователями FDA в 2015 году, спонсоры избегали упоминания 85% опасений FDA по поводу безопасности и эффективности при публичном объявлении об отклонении их заявки. Более того, когда FDA призывает к проведению нового клинического исследования безопасности или эффективности, эта критически важная информация не раскрывается примерно 40% раз. Уроки, извлеченные из случаев не одобрения, также не распространяются внутри отрасли, что приводит к тому, что компании неоднократно совершают аналогичные ошибки. – Читать далее: FDA стремится к радикальной прозрачности, публикуя полные ответные письма –

{kind=link}