
How effective is ELEVIDYS (delandistrogene moxeparvovec-rokl) gene therapy, which has been introduced to the pharmaceutical market, in relation to its cost of 3 million American Dollars? The clinical trials review of Elevidys Gene Therapy will be the main topic of this essay. We will assess the cost and effectiveness of this gene therapy, which was developed to treat Duchenne muscular dystrophy. Is Elevidys Cost Worthy Compared to Clinical Trials?
Table of Contents
Overview of Clinical Trials
The Food and Drug Administration (FDA) authorized a revolutionary gene therapy in 2023 and 2024 for the rare but fatal childhood-onset condition Duchenne muscular dystrophy.

Only youngsters aged 4-5 who could walk (ambulatory) were eligible for the 2023 permission.
The 2024 ruling broadened that clearance to cover conventional approval for ambulatory patients with the condition and a verified mutation in the DMD gene who are 4 years of age or older.
Additionally, the FDA established accelerated approval for people 4 years of age and older who depend on wheelchairs.
Neither approval was backed by appropriate clinical trial results showing that children with DMD retained or restored gross motor ability, nor by positive FDA scientific evaluations.
An FDA director disregarded the agency’s scientific staff, resulting in both approvals.
The two poorly thought-out approvals are discussed in this paper along with the reasons why FDA leaders should not follow their pattern in the future. It also offers suggestions for how the FDA can alter the way it handles contentious regulatory decisions.
How Elevidys Got Approved by FDA
The two Phase 3 randomized clinical trials of delandistrogene moxeparvovec-rokl (Elevidys) as a gene replacement cure for Duchenne muscular dystrophy (DMD) were reported in Nature Medicine on October 29, 2024. [1] The clinical efficacy of this medication has not been conclusively proven for two reasons, according to that report.
Both trials did not achieve their primary endpoints for clinical benefit. The clinical benefit was evaluated using a standardized scale that measures the trajectory of gross motor performance, including standing, walking, jumping, and head lifts.
Secondly, alternative endpoints that were marginally favorable—ultimately employed by the Food and Drug Administration (FDA) to support approval—could not be verified through statistical testing.
Additionally, the primary alternative outcome was the levels of micro-dystrophin protein, a biomarker that recently did not show clinical relevance in another DMD gene therapy trial. [2]
When Was Elevidys Approved?
Despite the limited clinical evidence, on June 22, 2023, delandistrogene moxeparvovec-rokl received FDA approval as the first gene replacement therapy for Duchenne muscular dystrophy (DMD), specifically for ambulatory children aged 4 to 5 years. [3]
The approval was obtained through the FDA’s accelerated approval program. [4]
On June 20, 2024, the FDA broadened its approval to encompass traditional authorization for ambulatory individuals aged 4 years and older diagnosed with the disease and possessing a confirmed mutation in the DMD gene. The agency has also granted accelerated approval for individuals aged 4 years and older who are dependent on wheelchairs. [5]
A concerning statement was noted in the FDA’s clinical review document pertaining to the expanded approval action: [6]
Dr. Peter Marks, Director of the Center for Biologics Evaluation and Research (CBER), is approved the application by overruling the recommendation of the review team.
Delandistrogene moxeparvovec-rokl is now approved for marketing in the United States. The therapy has recently been priced at more than $3 million per cure. [7]
Duchenne Muscular Dystrophy (DMD) is a rare, progressive, and fatal condition characterized by a scarcity of effective cure alternatives.
Duchenne Muscular Dystrophy (DMD) mutations are primarily recessive and X-linked, resulting in a predominant impact on males. The incidence of birth in males is roughly 1 in 3,600. [Read More: What is DMD?]
These mutations result in the degradation of skeletal and cardiac muscle, which manifests in early childhood, despite otherwise normal musculoskeletal development. Individuals with a DMD genotype typically lose ambulation by early adolescence and have a life expectancy that often concludes in their thirties.
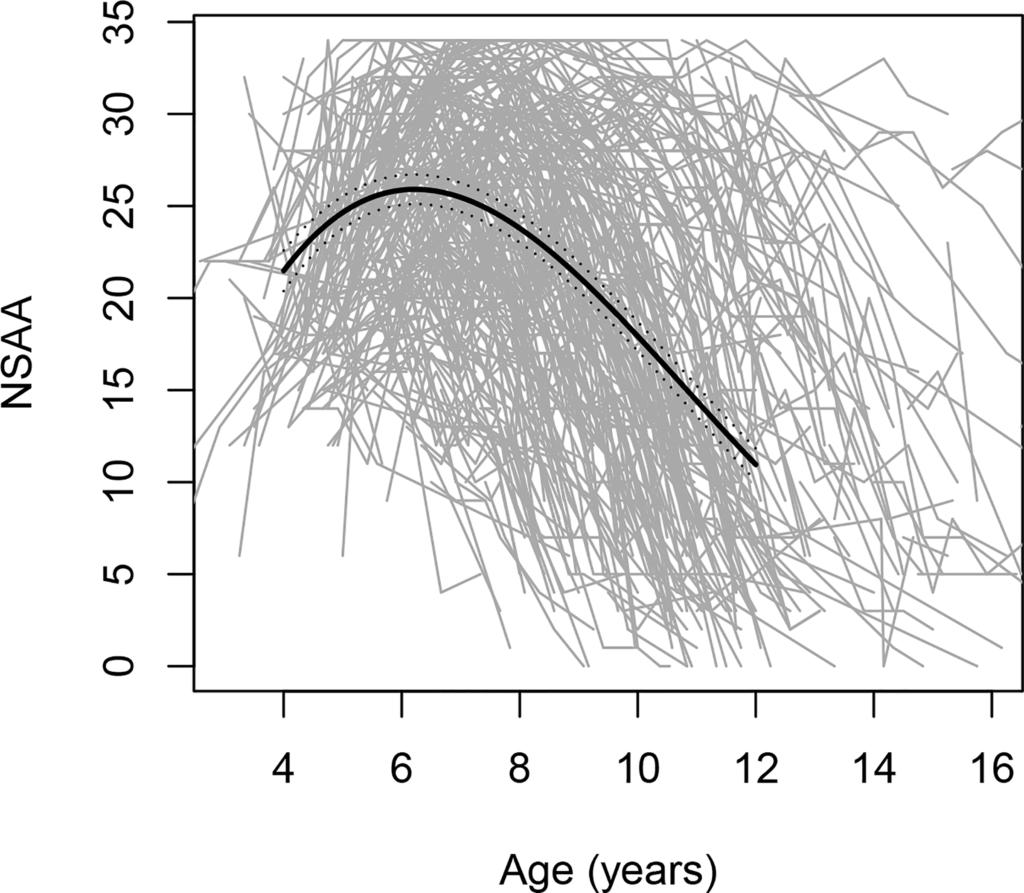
The disease course is heterogeneous, as evidenced by FDA data indicating significant variability in motor function trends among boys with DMD aged 4-16 years (see figure).
Figure. Changes in North Star Ambulatory Assessment (NSAA) scores, a gross motor performance scale, with age; includes 395 individual trajectories, each depicted by a grey line.
Sarepta’s DMD Cures
Delandistrogene moxeparvovec-rokl represents the inaugural disease-modifying therapy for the majority of Duchenne muscular dystrophy (DMD) cases; however, the FDA had earlier sanctioned four medications for less common variants of DMD.
Eteplirsen (Exondys 51) is a drug under scrutiny, designed to address specific DMD mutations through the use of antisense oligonucleotides that enable targeted “exon skipping,” thereby bypassing mutated regions in gene-to-protein expression. [8]
There are five FDA-approved cures for DMD, including delandistrogene moxeparvovec-rokl, all of which utilize truncated forms of the essential muscle protein dystrophin (as detailed below). [9]
All of these cures received approval via the FDA’s accelerated approval program. Despite being utilized in patients for several years, the majority of these cures have not shown significant clinical advantages over placebo.
Additionally, four of the cures, including delandistrogene moxeparvovec-rokl, are produced by a single company, Sarepta Therapeutics. [10]
Dr. Robert Califf, the FDA commissioner during the approval of both eteplirsen and delandistrogene moxeparvovec-rokl, expressed ongoing concerns regarding Sarepta’s DMD products in 2024, stating, “Sarepta’s like a curse to me.” [11]
Elevidys’s Mechanism of Action
The expression of a gene that codes for the protein dystrophin is significantly reduced or silenced in mutations that cause DMD.
Many biochemical components of the protein cross the membranes of muscle cells. Additionally, it serves as a shock absorber, keeping muscles strong in the face of potential deterioration brought on by frequent contraction-expansion cycles. [12]
Theoretically, DMD can be lessened or even reversed by encouraging dystrophin expression in the muscle cells of those who have it. The delandistrogene moxeparvovec-rokl, whose active component is a modified RNA sequence that encodes for a shortened variant of dystrophin known as “micro-dystrophin,” was developed in response to this notion.
Micro-dystrophin is about 30% of the size of unmutated dystrophin. The shortened protein seen in patients with the milder Becker type of muscular dystrophy served as the basis for the invention of the micro-dystrophin encoded by the delandistrogene moxeparvovec-rokl.
Because the RNA sequence had to be bundled into viral capsids in order to be delivered into the body, the dystrophin gene had to be truncated. Full dystrophin RNA surpasses the capacity limit of such capsids.
The delandistrogene moxeparvovec-rokl was created using a complex bioengineering technique that results in a protein that, at most, would only partially restore dystrophin function throughout the body. The effectiveness of this intricate therapy has not yet been shown in clinical trials.
Accelerated Approval of 2023
Forty-one ambulatory children with DMD, ages 4 to 7, participated in the pivotal trial for the first approval of delandistrogene moxeparvovec-rokl. They were randomized 1:1 to receive the medicine or a placebo.
The main predetermined clinical outcome was the change in gross motor performance over a 48-month period as measured by a 17-item standardized scale; patients treated with delandistrogene moxeparvovec-rokl did not significantly enhance their performance as compared to the control group. [13]
Based on the surrogate endpoint of micro-dystrophin expression in muscle tissue 12 weeks following the gene therapy cure, Sarepta, the sponsor, requested fast approval due to the unfavorable clinical trial outcome.
Despite the fact that microdystrophin expression was noted at 12 weeks, there was no correlation between microdystrophin levels and the ensuing muscle function. Consequently, the FDA scientific reviewers came to the conclusion that there was no therapeutic benefit to treating DMD from the delandistrogene moxeparvovec-rokl trial. According to the FDA clinical review from 2023: [14]
Therefore, even for a small population, such as ambulatory patients with DMD aged 4 to 5 who have a confirmed DMD mutation in their DMD gene, these data are insufficient to support the expression of ELEVIDYS micro-dystrophin as a surrogate endpoint “reasonably likely to predict clinical benefit” for Accelerated Approval of ELEVIDYS.
The FDA’s Center for Biologics Evaluation and Research director disregarded the agency’s scientific reviewers and decided to award fast approval in ambulatory 4- and 5-year-olds with DMD in spite of this harsh criticism.
Data from 8 of the 41 patients served as the primary basis for the decision. Additionally, the sponsor was “required as a condition of accelerated approval” to finish a second randomized trial, the results of which were expected by fall 2023, according to the ruling. [15]
Expanded Approval in 2024
Sarepta said in a news release dated October 30, 2023, that the primary endpoint of the second pivotal randomized trial of the delandistrogene moxeparvovec-rokl was not met.
In that placebo-controlled study, 125 patients between the ages of 4 and 7 were randomly assigned 1:1 to receive either a placebo or the delandistrogene moxeparvovec-rokl. [16]
52-week change on the same muscular function scale score used in the previous experiment was the main endpoint. Only a 0.65-point average difference between the medicine and placebo on the standardized gross motor functional scale (which has a maximal range of 34 points) was found, making the results not significant (p=0.25).
The FDA should have stopped approving delandistrogene moxeparvovec-rokl there. To the 2023 approval, the maker instead emphasized secondary outcomes that they said produced “robust, statistically significant results…[that] support an efficacy supplement.”
The FDA scientific reviewers were harshly dismissive of this new findings. In particular, the 10-meter walk/run tests and the time it took to get up from a floor position were two important secondary outcomes.
Delandistrogene moxeparvovec-rokl subjects performed on these activities on average 0.5 seconds faster than placebo subjects, who took 3.5 and 4.9 seconds at baseline, respectively.
These findings, which were neither prespecified nor statistically corrected for the multiple data analyses, “cannot support effectiveness of ELEVIDYS,” according to the FDA reviewers.
The data, according to the FDA reviewers, were also “misleading and cannot guide any stakeholder — including patients, family members and caregivers, and prescribers — in making informed decisions about the potential benefits of cure with ELEVIDYS.” [17]
However, in June 2024, the CBER Director overruled his team and approved the delandistrogene moxeparvovec-rokl for the second time, despite the FDA scientists’ unfavorable reviews. [18]
As was previously mentioned, the second approval expanded and strengthened the permission to include typical approval in ambulatory patients with the condition who are 4 years of age or older and have a verified mutation in the DMD gene. Additionally, accelerated permission for wheelchair-dependent people aged 4 and up was incorporated in the second approval. [19]
The surrogate indication of connection between micro-dystrophin protein levels and the time to rise from laying on the floor data was a major factor in the CBER Director’s 2024 decision memo.
This link, however, was neither visually (graphically) convincing or statistically significant (p=0.1388) due to the shallow slope (-0.012 seconds for every percent change in the muscle’s micro-dystrophin level).
Furthermore, a large number of data points deviated significantly from the estimated regression line.
Because just 25% of the individuals in the second pivotal trial were included in this analysis, FDA experts had explicitly noted that the results should be “interpreted with caution” when evaluating the micro-dystrophin dose-response data. As a result, despite the scientific justification for using micro-dystrophin gene insertion to treat DMD, clinical trials have not shown that the gene therapy has any positive effects. [20]
Indeed, “significant amounts” of micro-dystrophin expression were seen in the muscles of DMD patients in a recent Phase 3 trial of a Pfizer-developed DMD gene therapy, but there was no commensurate clinical improvement. According to reports, Pfizer has stopped working on their cure. [21]
Family Comments
Similar worries have been voiced by those who are directly impacted by DMD. Concerns regarding the safety and efficacy of delandistrogene moxeparvovec-rokl led one parent to write in September 2024 that they had chosen not to pursue therapy for their child. [22]
In an online video, a different parent criticized Sarepta for creating a cure that was insufficient and will soon become outdated. In response, a parent support group that was hosting the video was accused of removing the delandistrogene moxeparvovec-rokl video-critique from its website after Sarepta Therapeutics threatened to stop funding the group. [23]
Is Elevidys Cost Worthy Compared to Clinical Trials?
Considering the outcomes of the clinical trials, is the 3 million USD price tag for the Elevidys gene therapy reasonable? [Read More: Duchenne Muscular Dystrophy: Treatment & Cost]
Considering the amount of dystrophin produced in the body after Elevidys use and the North Star Ambulatory Assessment (NSAA) scores examined in clinical trials, 3 Million USD is very exorbitant for this treatment method.
Our call to Sarepta, the developer of Elevidys, and to Roche, the global marketing authority: Reduce this high price set for this commercial purpose immediately!
Learn More: Potential Upcoming New Gene Therapies for Duchenne Muscular Dystrophy
First Death Announcement
On March 18, 2025, Sarepta Therapeutics said that the first recorded fatality associated with their gene therapy for Duchenne muscular dystrophy, Elevidys, had occurred in a 16-year-old boy. [Read More]
Conclusion
A center director overruled the FDA’s scientific team, who were tasked with assessing the medicine, and the FDA approved delandistrogene moxeparvovec-rokl as a gene therapy for DMD.
Delandistrogene moxeparvovec-rokl’s two pivotal trials failed to meet their main clinical goals. Indicators used to rescue the delandistrogene moxeparvovec-rokl application, such as secondary endpoints (e.g., 10-meter walk/run tests) and the indirect surrogate indicator of micro-dystrophin levels, were problematic in and of themselves.
As a result, we think the FDA leadership has created a risky precedent that does not shield the public from being exposed to an ineffective medicine that is expensive and physically demanding. [24]
Learn More: Duchenne Muscular Dystrophy Market Grows: But Not All Families Have Access to Treatments
One way to react to the FDA’s ruling is to say that weak approval criteria are the issue and that stricter approval standards and post-marketing procedures are the solution. [25]
The FDA’s internal processes should be changed to make it much more difficult for individual officials in leadership roles to override the consensus assessment of agency scientists assigned to a regulatory decision, even though we agree that stricter approval standards and post-marketing requirements are required.
According to Califf, the FDA Commissioner at the time of the two delandistrogene moxeparvovec-rokl approval decisions, those who oppose the decision have a “very simplistic view of clinical evidence” about the difficulties in developing therapies for serious and uncommon disorders. [26]
We don’t agree with that evaluation. The FDA should enhance its decision-making process for new pharmaceuticals, whether they are for common or uncommon illnesses, as a result of the approval of delandistrogene moxeparvovec-rokl.
Learn More: DMD WarrioR’s Turkish Representative Shares His Views on Elevidys Gene Therapy
In a pre-leaving interview, Califf also discussed the decision, pointing out that families with such severe and uncommon illnesses were understandably yearning for any hope, and that as a political appointee, he was also reluctant to overturn the decisions made by career FDA employees. While these factors are significant, they are not enough to support the FDA’s decision to approve the delandistrogene moxeparvovec-rokl. [27]

{kind=link}