









 FDA en 2019")
La Administración de Alimentos y Medicamentos publicó más de 200 cartas que envió a las empresas después de que sus medicamentos fueran rechazados el jueves, llamando la atención sobre una parte del proceso de revisión de medicamentos que a veces se pasa por alto. Una de estas cartas de rechazo fue enviada para Vyondys 53 (golodirsen), un fármaco fabricado por Sarepta Therapeutics y desarrollado para la terapia de omisión del exón 53.
La agencia se centró únicamente en las cartas a los fabricantes cuyos productos finalmente fueron aprobados, la mayoría de los cuales ya se habían publicado a lo largo de los años. Al reunir todas las cartas en un solo lugar, la agencia afirmó que la actividad formaba parte de un esfuerzo más amplio para promover la transparencia.

Tabla de contenido
Detalles de la carta de rechazo FDA sobre Vyondys 53
El FDA emitió su carta de rechazo de 2019 para Vyondys 53 (golodirsen), un tratamiento de omisión del exón 53 para la distrofia muscular de Duchenne. El FDA afirmó que, dado el “pequeño aumento en los niveles de distrofina acortada” proporcionado por el tratamiento, el beneficio clínico de Vyondys 53 probablemente sería “proporcionalmente pequeño”.
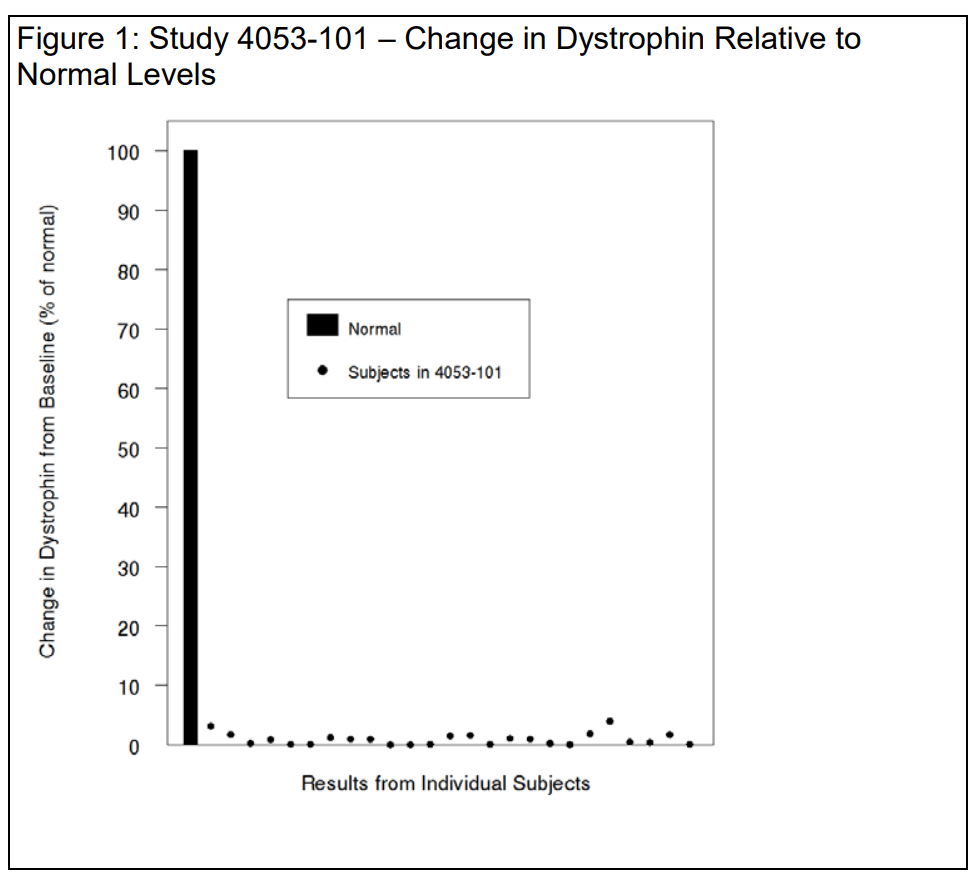
El nivel de distrofina aumentó 9 partes por mil
La carta de rechazo de FDA para Vyondys 53 (golodirsen) notó un aumento muy pequeño en los niveles de distrofina después del uso del medicamento.:
En los 25 pacientes evaluables del estudio, el nivel medio basal de distrofina, determinado mediante Western blot, fue de 0,10 ± 0,07 (porcentaje del valor normal; media ± DE). En la semana 48, el nivel medio de distrofina fue de 1,02 ± 1,03 (porcentaje del valor normal; media ± DE), lo que corresponde a un aumento medio del 0,92 ± 1,01 % del valor normal.
Los cambios individuales en la distrofina se muestran a continuación en la Figura 1, utilizando una escala normal (100%) para contextualizar los datos y evitar la exageración de la magnitud del efecto. Observa que los aumentos medios en la distrofina truncada son similares en respuesta a golodirsén y eteplirsén en pacientes con mutaciones susceptibles de omisión del exón 53 y del exón 51, respectivamente, con aumentos medios absolutos de 0,9%, es decir, 9 partes por mil. Estamos de acuerdo.
Si se acepta la premisa de que es razonablemente probable que un pequeño aumento de distrofina truncada, del orden de 9 partes por mil, prediga un beneficio clínico, parece razonable suponer que el beneficio clínico sería proporcionalmente pequeño.
Efecto clínico de Vyondys 53
Los datos de la caminata de 6 minutos del Estudio 4053-101 no se analizan en este documento, pero muestran una pérdida progresiva de la función física en prácticamente todos los niños. Además, no existe correlación entre el mantenimiento del rendimiento físico y la magnitud de la producción truncada de distrofina, lo que sugiere que, si bien el golodirsen tiene un efecto clínico, su magnitud es pequeña.
Efectos secundarios graves de Vyondys 53, según el FDA
En diciembre de 2019, el FDA autorizó Vyondys 53 de Sarepta como la primera opción terapéutica dirigida para pacientes con DMD susceptibles a la omisión del exón 53. Sin embargo, el medicamento había sido rechazado por el FDA cuatro meses antes en una CRL firmada por Ellis Unger, quien en ese momento era director de la Oficina de Evaluación de Medicamentos del Centro de Evaluación e Investigación de Medicamentos.
El FDA declaró en su justificación del rechazo que, considerando el “pequeño aumento de distrofina truncada” que el tratamiento provocó, el efecto terapéutico de Vyondys 53 probablemente sería “proporcionalmente pequeño”. Unger continuó explicando que no existía correlación entre la cantidad de distrofina producida por Vyondys 53 en los niños y su rendimiento físico, y que “prácticamente todos” los niños examinados después de recibir el fármaco mostraron una pérdida creciente de la función física.
Como si los moretones no fueran suficientes, Unger enumeró una larga lista de problemas de seguridad que los usuarios de Vyondys 53 tuvieron que afrontar. Estos incluían "toxicidad renal" e "infecciones graves relacionadas con la administración del fármaco". Afirma que ambas "tienen el potencial de ser mortales", siendo la última "difícil o imposible de controlar".
Dadas las dos muertes recientes por insuficiencia hepática grave relacionadas con la terapia génica Elevidys de Sarepta, esta segunda advertencia es especialmente preocupante. Como señaló Unger en la carta, ambas vías se han vinculado con daño hepático, a pesar de que Vyondys 53 es un oligonucleótido antisentido (ASO), a diferencia de una terapia génica basada en virus adenoasociados (AAV), como Elevidys.
Leer más: Carta de rechazo de Vyondys 53 FDA
¿Cómo se aprobó Vyondys 53?
Para abordar las preocupaciones planteadas por el regulador en la CRL, Sarepta presentó posteriormente una apelación y se reunió con el FDA. Esto finalmente resultó en que el entonces director de la Oficina de Nuevos Medicamentos del FDA, Peter Stein, quien dejó la agencia en abril, aprobara Vyondys 53.
Las compañías farmacéuticas no publican toda la información
“Durante demasiado tiempo, los desarrolladores de fármacos han estado jugando a las adivinanzas al navegar por el FDA”, dijo el Comisionado del FDA, Dr. Marty Makary, MPH. “Tanto los desarrolladores de fármacos como los mercados de capitales buscan previsibilidad. Por eso, hoy estamos un paso más cerca de proporcionársela, con el objetivo final de brindar curas y tratamientos eficaces a los pacientes más rápidamente”.
Dado que el FDA históricamente se ha abstenido de publicar las CRL de las solicitudes pendientes, los patrocinadores a menudo tergiversan la justificación de su decisión ante las partes interesadas y el público. Según un análisis realizado en 2015 por investigadores del FDA, los patrocinadores evitaron mencionar las preocupaciones del FDA sobre la seguridad y la eficacia al anunciar públicamente la no aprobación de su solicitud. Además, cuando el FDA solicita un nuevo ensayo clínico para evaluar la seguridad o la eficacia, esa información crucial no se divulga aproximadamente el 40% de las veces. Las lecciones aprendidas de las no aprobaciones tampoco se comparten dentro de la industria, lo que lleva a las empresas a cometer errores similares repetidamente. – Leer más: FDA adopta una transparencia radical al publicar cartas de respuesta completas –

{kind=link}